クオリティチェックの代表的なプログラムとして FastQC がある。FastQC は Java で書かれているため、OS を問わずにダウンロードすればすぐに利用できる。Linux 上では GUI 画面が利用できるに加え、コマンドラインからも利用でき、非常に便利なプログラムである。
FastQC の使い方
ここではコマンドラインから FastQC を利用する方法を示す。FastQC がインストールされれば fastqc コマンドが使えるようになる。基本的には fastqc コマンドのあとにチェック対象のファイルを与えればよい。
チェック結果のレポートを指定のディレクトリに保存するには -o オプションで指定する。また、リードが長くなると、3' 末端では十数塩基が束ねて解析される。そのため、3' 末端の解析結果が曖昧となることがある。これを防ぐためには --nogroup オプションを付ける。
例として、SRR610711.fastq に対してクオリティチェックを行い、その結果を reports ディレクトリに保存する場合は以下のようにする。fastqc を実行する前に makedir を利用して予め reports ディレクトリを作成しておく。
mkdir reports
fastqc --nogroup -o ./reports SRR610711.fastqコマンドが実行し終えると、指定した出力ディレクトリにはレポートが保存される。reports/SRR032116_fastqc/fastqc_report.html のファイルをブラウザーで開けば、レポートが見られる。レポートには以下のような情報が書かれている。これらの情報を参照し、FASTQ ファイルにある不自然なリードやクオリティの低いリードを取り除く。
--nogroup を付けるか付けないかの実行結果は以下のように違いがある。
--nogroup を付ける場合、3' 末端のリードについても解析される。(画像が横長になってしまう。ウェブページの幅の都合上、ここでは縮小して表示させている。)

--nogroup を付けない場合は、3' 末端のリードが束ねて解析される。
FastQC レポートの見方
詳細な FastQC レポートの見方については本家の FTP サイトに譲る。以下に、レポートの見方を簡単に紹介する。レポート結果は、次のようにいくつかの項目で表示される。良い、まあまあ、悪いの 3 段階で評価され、それぞれ緑、黄、赤でマークされる。ただし、これらの項目は FastQC が判定したものであり、それぞれのデータの特徴を考慮した結果となっていない。従って、赤とマークされていても、それを説明できる生物的な理由があれば、適切と判断してよい。
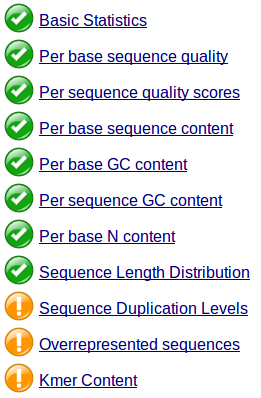
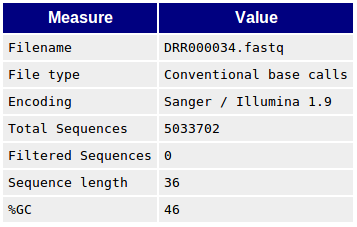
Basic Statistics
レポートの総括が記載されている。クオリティチェックした FASTQ ファイルの名前、リード数、リード長などの基本情報を確認できる。
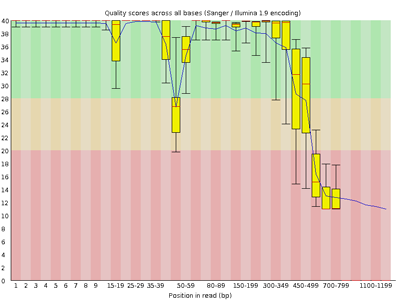
Per base sequence quality
リードの各位置におけるクオリティスコアの分布が示されている。横軸がリード上の位置を表し、縦軸はクオリティスコアを表す。スコアの分布はボックスプロットによって描かれる。スコアの下側四分位点(1/4 分位点)が 10 未満または中央値が 25 未満の位置が存在する場合 Warning とされる。また、スコアの下側四分位点(1/4 分位点)が 5 未満または中央値が 20 未満の位置が存在する場合 Warning とされる。
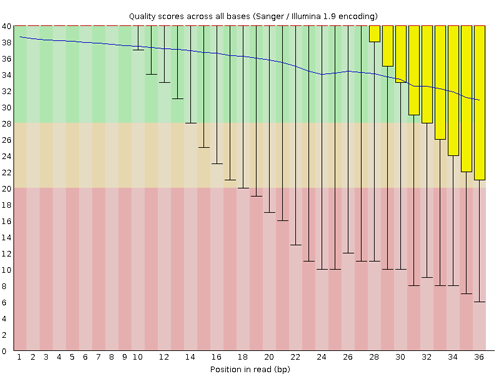
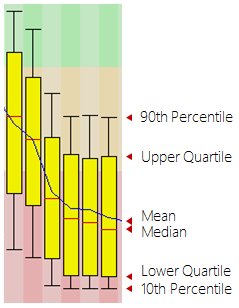
レポートの画像のボックスプロットは、次のようにそれぞれ 90% 百分位点、第 3 四分位点、中央、平均、第 1 分位点、10% 百分位点を表す。
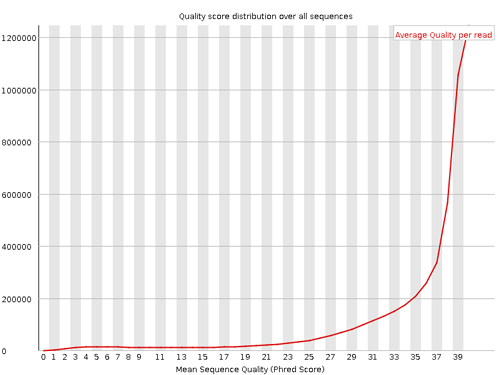
Per sequence quality scores
各クオリティスコアを持つリードがどれぐらい存在するかを示している。横軸がクオリティスコアを示し、縦軸はリード数を示す。スコアの高いところにリード数が多くなることが相応しいと考えられる。リード数の最も多いスコアが 27 未満ならば Warning とされる。また、20 未満ならば Failure とされる。
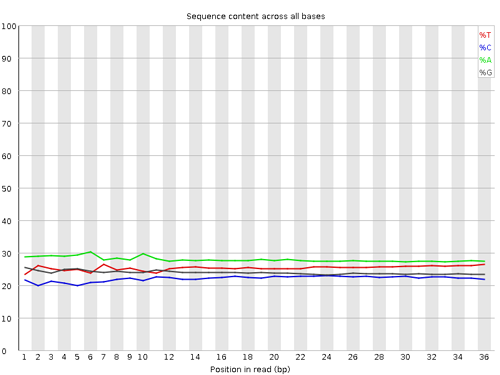
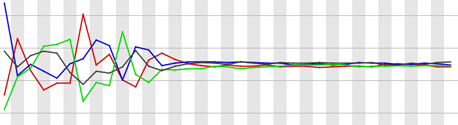
Per base sequence content
リードの各位置における塩基の出現頻度が示されている。シーケンシングされる断片は mRNA からランダムに断片化されたならば、各位置における塩基の出現頻度が等しいと期待される。つまり、出現頻度を表す折れ線グラフはほぼ横軸に平行な直線になると期待される。A と T、または G と C の差が 10% 以上ならば Warning とされる。また、20% 以上ならば Failure とされる。
次の図に見られるように、一時期昔によく使われた試料調整方法で調整したサンプルの RNA-seq データでは、最初の 13 残基がほかの比べ出現頻度がランダムでないように見えるが、これは試料調製する際に利用した DNase I に由来するもので、シーケンシングエラーではない。
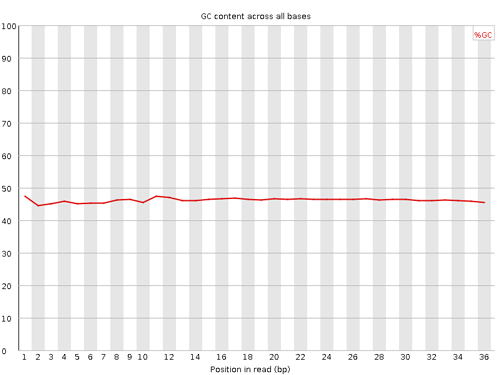
Per base GC content
リードの各位置における GC 含量が示されている。塩基配列が無作為にシーケンシングされているならば、各位置において GC 含量がほぼ同じであると期待される。つまり、折れ線グラフは横軸に平行すると期待される。GC 含量の平均から 5% 以上離れている位置があれば Warning とされる。また、10% 以上離れている位置があれば Failure とされる。
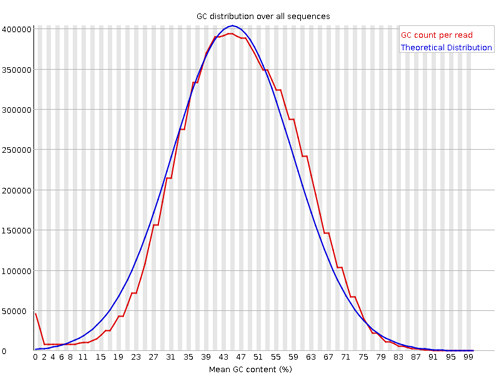
Per sequence GC content
リードの GC 含量の分布が示されている。リードに含まれる GC 含量は一般に正規分布に従うとされている。正規分布と比較し、その残差が 15% 以上ならば Warning とされる。また、30% 以上ならば Failure とされる。
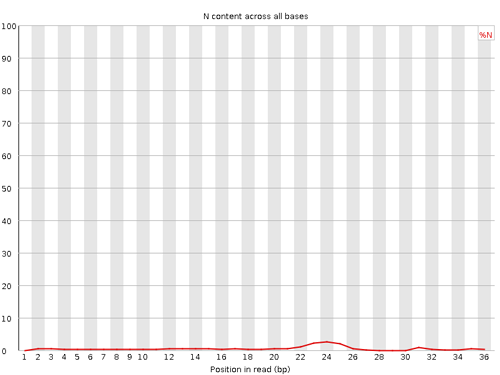
Per base N content
リードの各位置に現れる N の割合が示されている。N が 5% を上回る位置が存在するとき Warning とされる。また、20% を上回る位置が存在するとき Failure とされる。
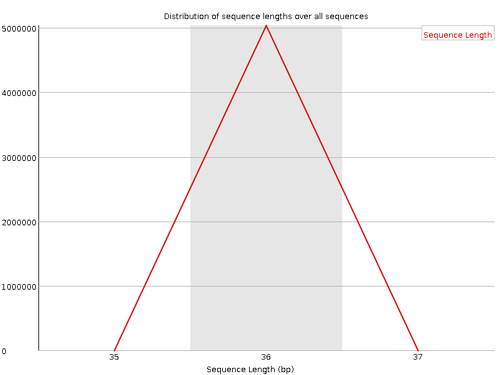
Sequence Length Distribution
リード長の分布が示されている。シーケンサーの種類によって、リードがすべて同じ長さで出力されるものもあれば、ばらばらな長さで出力される場合もある。また、クオリティコントロールを行ったならば、処理後にリード長が不揃いになる場合も考えられる。リードの長さがすべて同じでなければ Warning とされる。また、リードの長さがすべて 0 ならば Failure とされる。
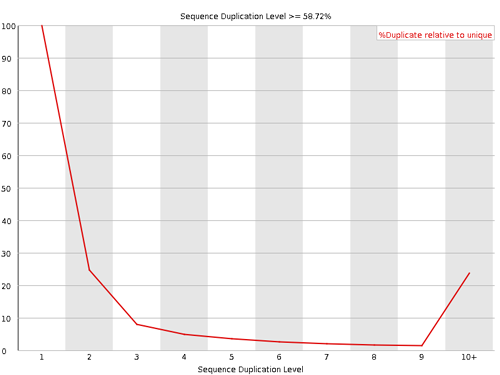
Sequence Duplication
リードの重複数を示している。サンプルを調整するとき mRNA の断片化を行う際に、ランダムに行われるので、リードの塩基構成もランダムである。すなわち、同じサンプルの中に全く同じ塩基配列を持つリードは 2 つ以上存在しないと考えられる。例えカバレッジが大きくても、mRNA の断片化がランダムに行われることを考慮すれば、重複リードが存在しないと考える。重複リードが多いときは、アダプターによるコンタミ、試料自体にリピート配列が多い、などが原因として考えられる。グラフの縦軸は割合を表す。重複していないリード数(duplication level 1)を 100% とし、他は duplication level 1 と比べたときの割合として出力される。下図では level 2, 3, 10+ の割合を相加するとほぼ 59% になる。これがグラフのタイトルとなている(58.72%)。
Overrepresented sequences
重複数の多いリードが表示される。例えば、試料調整のときにポリ A をうまく取り除けなかったりする場合は、以下のようなリードが overrepresented として表示される。

Kmer Content
リードから観測される K-mer が示されている。
インストール
FastQC は Java でかかえれているプログラムであり、ほとんどのシステムでダウンロードしてくるだけで使えることができる。FastQC はBabraham Instituite/ からダウンロードできる。

プログラムをダウンロードしてから、その zip を展開すると FastQC ディレクトリが作成される。このディレクトリ中の fastqc ファイルがあり、このファイルを実行ファイルに変更する。
cd FastQC
chmod +x fastqcFastQC の使い方(GUI)
パスを通してあれば、ターミナルウィンドウで fastqc をタイプするだけで利用できる。
fastqcターミナルとは別の FastQC アプリケーションのウィンドウが立ち上がる。FastQC のウィンドウから「File」、「Open」の順をクリックし、解析を行いたいファイルを選べば、自動的に解析が行われ、解析結果がウィンドウ上に表示される。
解析結果が以下のように表示される。
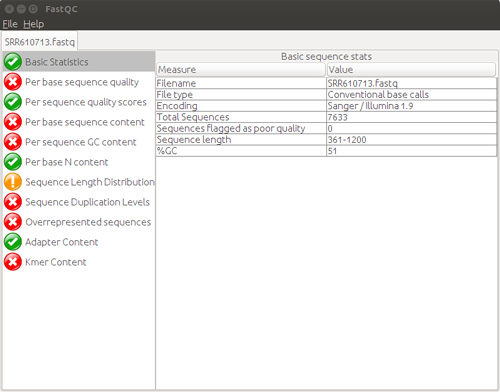
FastQC の使い方(CUI)
fastqc コマンドの後に解析対象のファイルを指定すれば、FastQC はコマンドラインモードで動作する。
fastqc SRR610713.fastq解析結果を特定のディレクトリに保存する場合は -o オプションを指定する。
fastqc -o ./reports SRR610713.fastqFastQC には -o の他にもいくつかのオプションが用意されている。「fastqc -h」を実行することでこれらのオプションを確認できる。
| オプション | 動作 |
-o |
解析結果の保存先ディレクトリを指定する |
--nogroup |
リードが 50 bp よりも長い場合、FastQC は 3' 末端にある塩基を束ねて解析を行う。--nogroup を付けることによってこれらの塩基を束ねずに 1 ポジションごとに処理する。 |
-f |
入力ファイルのフォーマット。デフォルトは fastq である。このほかに bam、sam にも対応している。 |
-t |
使用しているスレッド数(CPU 数)。 |
-c |
コンタミが想定されている配列がある場合、それらコンタミ配列をタブ区切りのテキストファイルに保存してから、-c オプションで指定する。フィアルは以下のように、「配列名、タブ、配列」の順で 1 行ずつ記述する。
Contaminant_seq_1 AACGAGTCCC Contaminant_seq_2 AACCAGTCTT |
-a |
アダプター配列が使われている場合、そのアダプター配列をタブ区切りテキストに保存してから、-a オプションで指定する。ファイルは以下のように、「アダプター名、タブ、配列」の順で 1 行ずつ記述する。
GenomicDNASeq ACACTCTTTCCCTACACGACGCTCTTCCGATCT NlaIII ACAGGTTCAGAGTTCTACAGTCCGACATG |
-k |
リードの Kmer を解析する際に必要な Kmer の長さを指定する。2 から 10 の間を指定する。デフォルトは 7 である。 |