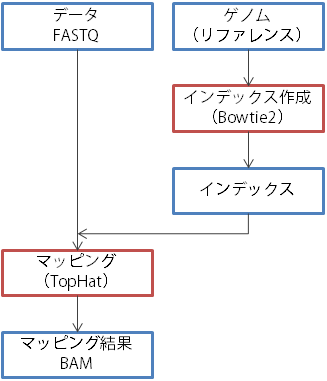
TopHat はマッピングプログラムの一つで、長めのリードのマッピングによく用いられる。シーケンサーから出力されるリードは長さ 100 bp 以上のものもある。これらのリードはエクソンの長さを超えているため、転写産物からなる(イントロンを含まない)リードをイントロンを含むリファレンスゲノムに、うまくマッピングすることはできない。
そこで TopHat は長いリードを短いリードに断片化した上でマッピングを行い、1 個のリードが複数のエクソンからなる場合もマッピングできる。
このページでは SRR384905 と SRR384906 をサンプルデータとして、TopHat を利用してマッピングする例を示す。これらのデータはショウジョウバエの RNA-seq の結果である。リードは paired-end リードとなっているため、サンプルデータを FASTQ 形式に変換すると SRR384905_1.fastq、SRR384905_2.fastq、SRR384906_1.fastq、SRR384906_2.fastq の 4 つのファイルができる。
TopHat を利用したマッピングの流れは、おおよそ以下のようになっている。
公共データベースからリファレンスゲノムをダウンロードする
ショウジョウバエの全ゲノムデータは公共データベースである Ensembl でダウンローできる。しかし、全ゲノムをダウンロードしても、どの領域が遺伝子でどの領域がそうではないかのアノテーション情報が含まれていない。そのため、(マッピングには必要はないが、)全ゲノムの他にアノテーションデータも合わせてダウンロードすると便利である。
例えば、ショウジョウバエ関連のデータは Ensembl FTP で Fruitfly の項でダウンロードできる。

上図の赤い枠で囲まれたファイルをダウンロードする。
- Ensembl FTP ページのショウジョウバエの項目から、DNA(FASTA) 列にある FASTA ファイルをダウンロードする。その他に、cDNA、CDS や Protein sequence など様々な FASTA ファイルが用意されているが、ダウンロードするのはあくまで DNA(FASTA) の FASTA である。該当リンクをクリックすると、複数のファイルがリストアップされる。このうち、.toplevel.fa.gz で終わるファイルをダウンロードする(1 つだけあるはず)。このファイルが全ゲノム配列である。
※_sm.toplevel.fa.gz は利用しないので、ダウンロードする必要がない。 - 同様に、Ensembl FTP ページの Gene sets 列にある GTF リンクをクリックすると、アノテーションデータのダウンロードページに移動できる。.gtf.gz で終わるファイルをダウンロードする。(このファイルは、マッピング時に利用しないが、後々の解析がスムーズに進めるように予めダウンロードしておくとよい。)
これらは gzip 形式で圧縮されているため、ダウンロード後にこれらを展開する。
gunzip Drosophila_melanogaster.BDGP5.75.dna.toplevel.fa.gz
gunzip Drosophila_melanogaster.BDGP5.75.gtf.gz展開すると、リファレンスとなる全ゲノムファイル Drosophila_melanogaster.BDGP5.75.dna.toplevel.fa と アノテーションファイル Drosophila_melanogaster.BDGP5.75.gtf が得られる。
インデックスファイルを作成
TopHat は内部的に Bowtie2 を利用しているため、Bowtie2 向けのインデックスファイルを作成する必要がある。ここでは上でダウンロードしたリファレンスのインデックスを作成する。インデックスの名前を BDGP75 とする。
bowtie2-build -f Drosophila_melanogaster.BDGP5.75.dna.toplevel.fa BDGP75インデックスを作成し終えると、ディレクトリには 6 つのファイルが生成される。
- BDGP75.1.bt2
- BDGP75.2.bt2
- BDGP75.3.bt2
- BDGP75.4.bt2
- BDGP75.rev.1.bt2
- BDGP75.rev.2.bt2
マッピング
このサンプルは paired-end リードであるから、以下のように *_1.fasq と *_2.fastq を続けて指定する。-o のあとにマッピング結果を保存するためのディレクトリを指定する。サンプルデータは 2 つあるので、それぞれに対してマッピングを行う。
tophat -o SRR384905_mapped BDGP75 SRR384905_1.fastq SRR384905_2.fastq
tophat -o SRR384906_mapped BDGP75 SRR384906_1.fastq SRR384906_2.fastq マッピングが終わると、2 つのディレクトリ(SRR384905_mapped と SRR384906_mapped)が生成される。それぞれのディレクトリの中にマッピング結果とマッピング時のログが書かれている。
- accepted_hits.bam - マッピング結果
- align_summary.txt - マッピングレポート
- deletions.bed
- insertions.bed
- junctions.bed
- perp_reads.info
- unmapped.bam
- logs - 様々なログ
以上で得られた SRR384905_mapped と SRR384906_mapped の accepted_hits.bam ファイルを HTSeq などのプログラムを用いれば、カウントデータが得られる。
カウントデータの取得
HTSeq を利用すると BAM ファイルからカウントデータを取得することができる。この時に、どの領域が遺伝子領域なのかを HTSeq に教える必要があるため、上述した方法でダウンロードした GTF フォーマットのアノテーションファイル(Drosophila_melanogaster.BDGP5.75.gtf)を利用する。また、BAM ファイルは遺伝子名でソートする必要があるため、SAMtools を利用してソートしてから HTSeq に与える。
## SRR384905
samtools sort -on SRR384905_mapped/accepted_hits.bam tmp_prefix \
| samtools view -h - \
| htseq-count - Drosophila_melanogaster.BDGP5.75.gtf > SRR384905.count.txt
## SRR384906
samtools sort -on SRR384906_mapped/accepted_hits.bam tmp_prefix \
| samtools view -h - \
| htseq-count - Drosophila_melanogaster.BDGP5.75.gtf > SRR384906.count.txt最後に、各ディレクトリそれぞれに SRR384905.count.txt と SRR384906.count.txt ファイルが作成される。これらのファイルは以下の例のように、タブ区切りで、1 列目に遺伝子名、2 列目にカウントデータが記載される。
カウントデータが複数のファイルにまたがっているため、これらを join コマンドで結合する。また、上述のカウントデータが記載されいてるファイルのなかに、遺伝子のカウントデータの他に、アノテーションがないリード数やマップされてなかったリード数なども保存されている。前者の場合は ENSG から始まり、後者の場合は「__no_feature」や「__not_aligned」などとなっている。そのため、grep 文を利用して ENSG だけから始まるデータのみを取得する。
# カウントデータのみを tmp.count.txt (一時ファイル)に保存する
join -j 1 SRR384905.count.txt SRR384906.count.txt > tmp.count.txt
# ヘッダーを all.count.txt に書き入れる
echo "EnsemblID SRR384905 SRR384906" > all.count.txt
# カウントデータを all.count.txt に書き入れる
grep "^ENSG" tmp.count.txt >> all.count.txt
# 一時ファイルを削除する
rm tmp.count.txtReferences
- TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14(4):R36. PubMed Abstract
- TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009, 25(9):1105-11. PubMed Abstract
- Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10(3):R25. PubMed Abstract
- Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012, 9(4):357-9. PubMed Abstract