IGV を利用して BAM ファイルを視覚化する例を示す。マッピング結果として SAM ファイルが得られた場合、まず最初に、これを BAM ファイルに変換する。(BAM ファイルならばこの変換作業を行う必要はない。)
samtools view -bS align.sam > align.bamIGV に読み込ませるには、まず BAM ファイルを染色体順にソートし、インデックス付けを行う必要がある。
samtools sort align.bam align.sorted
samtools index align.sorted.bamコマンドが正しく実行されると、align.sorted.bam ファイルと align.sorted.bam.bai ファイルが生成される。
次に IGV を起動し、リファレンスゲノムを予め読み込んでおく。次に、メニューから「File > Load from File..」をクリックし、align.sorted.bam を選択して開く。IGV の左のフレーム読み込んだ BAM ファイルの名前が表示される。IGV のメインのフレームにはマッピング結果を見ることができる。以下のように、マップされたリードが見れない場合は、表示範囲を拡大すると見えるようになる。
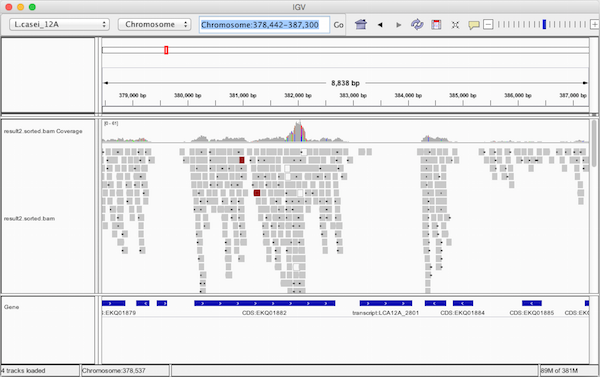
※メモリー不足で表示できない場合もある。特に、バーチャルマシン上で IGV を利用する際に、バーチャルマシンのメモリを十分に確保する必要がある。