BWA(Li et al, 2009, Li et al, 2010)はロングリードのマッピングを得意としている。ここで、トウモロコシのアセンブリゲノムの改良の目的でシーケンスされた DNA のロングリード(Jiao et al, 2017)を、BWA でトウモロコシ(Zea mays)のゲノムにマッピングしてみる。
DNA-Seq データ
ここでは PRJNA10769 の番号でアーカイブされている DNA-Seq のデータを利用する。この番号でアーカイブされているデータは HiSeq 2500 と PacBio RS II の 2 種類がある。今回は、PacBio RS II でシーケンシングされた single-end リードを利用する。全部で 9 ファイルあるが、BWA の実行テストであれば 2, 3 ファイルだけダウンロードすれば十分。
wget -O SRR2983660.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/000/SRR2983660/SRR2983660_subreads.fastq.gz
wget -O SRR2983661.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/001/SRR2983661/SRR2983661_subreads.fastq.gz
wget -O SRR2983662.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/002/SRR2983662/SRR2983662_subreads.fastq.gz
wget -O SRR2983663.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/003/SRR2983663/SRR2983663_subreads.fastq.gz
wget -O SRR2983664.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/004/SRR2983664/SRR2983664_subreads.fastq.gz
wget -O SRR2983665.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/005/SRR2983665/SRR2983665_subreads.fastq.gz
wget -O SRR2983666.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/006/SRR2983666/SRR2983666_subreads.fastq.gz
wget -O SRR2983667.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/007/SRR2983667/SRR2983667_subreads.fastq.gz
wget -O SRR2983668.fastq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/008/SRR2983668/SRR2983668_subreads.fastq.gzデータの一部(SRR2983660)に対して、クオリティとリードの長さを確認してみると、次のようになっている。60 kbp を超える非常に長いリードもあることを確認できる。
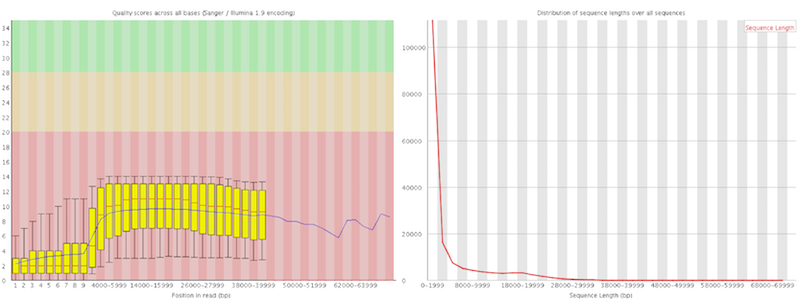
ダウンロードしたシーケンサーデータ(FASTQ ファイル)に対して、Trimmomatic を使って低クオリティリードの除去や短いリードの除去などを行う。フィルタリング後のファイルの名前を SRR2983660.clean.fastq.gz ように clean をつけて保存する。
SEQLIBS=(SRR2983660 SRR2983661 SRR2983662 SRR2983663 SRR2983664 SRR2983665 SRR2983666 SRR2983667 SRR2983668)
for seqlib in ${SEQLIBS[@]}; do
java -jar trimmomatic-0.38.jar PE -phred33 -threads 4 \
${seqlib}.fastq.gz ${seqlib}.clean.fastq.gz HEADCROP:35 MINLEN:100
doneゲノムの配列データ
トウモロコシ(Zea mays)のゲノムは Ensembl Plants で公開されている。このページから、ゲノム配列のデータをダウンロードする。
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-40/fasta/zea_mays/dna/Zea_mays.AGPv4.dna.toplevel.fa.gz
gunzip Zea_mays.AGPv4.dna.toplevel.fa.gzマッピングには必要としないが、マッピングされたリードを遺伝子領域ごとに集計するときにアノテーションファイルも必要である。このアノテーションファイルも(GFF または GTF)も合わせてダウンロードしておく。
wget ftp://ftp.ensemblgenomes.org/pub/plants/release-40/gff3/zea_mays/Zea_mays.AGPv4.40.gff3.gz
unzip Zea_mays.AGPv4.40.gff3.gzBWA によるマッピング
フィルタリングしたあとのリードデータ(SRR2960981_1.clean.fastq.gz と SRR2960981_2.clean.fastq.gz)とゲノムの配列データ(Zea_mays.AGPv4.dna.toplevel.fa)を準備できたので、次に BWA を使ってリードをゲノム配列にマッピングしていく。
BWA ではマッピングするときに、ゲノム配列から作成したインデックスを使用する。まず、このインデックスを作成する。インデックスの名前は任意につけることができる。ここではインデックスの名前を AGPv4 とした。
bwa index -p AGPv4 Zea_mays.AGPv4.dna.toplevel.faインデックスを作成し終えると、AGPv4.amb、AGPv4.ann、AGPv4.bwt、AGPv4.pac、AGPv4.sa の 5 ファイルが生成される。これらのファイルが BWA が使用するインデックスとなる。
次に、作成したインデックスを利用してリードをゲノム配列にマッピングしていく。ここで使用するリードデータは非常に長いリードとなっているため、1 ファイルをマッピングするのに数日かかる場合もある。BWA を試すのが目的であれば、リードデータからサブセットを作って、BWA を適用するとよい。BWA によるマッピング結果は SAM ファイルに保存される。
SEQLIBS=(SRR2983660 SRR2983661 SRR2983662 SRR2983663 SRR2983664 SRR2983665 SRR2983666 SRR2983667 SRR2983668)
for seqlib in ${SEQLIBS[@]}; do
bwa mem -t 4 TAIR10 ${seqlib}.clean.fastq.gz > ${seqlib}.sam
doneここで生成した SAM は、必要に応じて samtools で BAM に変換することができる。例えば、SRR2983660.sam をソートしながら BAM ファイルに変換するには次のようにする。
samtools sort -O bam -o SRR2983660.sorted.bam SRR2983660.sam
samtools index SRR2983660.sorted.bamReferences
- Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25(14):1754-60. DOI: 10.1093/bioinformatics/btp324
- Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010, 26(5):589-95. DOI: 10.1093/bioinformatics/btp698
- Improved maize reference genome with single-molecule technologies. Nature. 2017, 546(7659):524-7. DOI: 10.1038/nature22971