Bowtie2 は Burrows-Wheeler 法と Dynamic Programming 法を取り入れたマッピングプログラムで、マッピング速度が非常に速く、ギャップをも許容できる。Bowtie2 は主に 50bp-100bp 前後のリードのマッピングに用いられる。
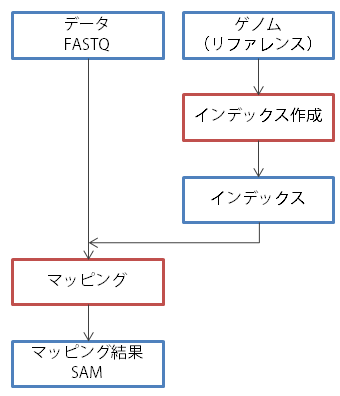
Bowtie2 を利用してリードをリファレンスゲノムへマッピングするには、おおよそ左図のような流れで行う。まず、公共データベースからリファレンスとなるゲノム配列をダウンロドして、インデックス化する。次に、FASTQ ファイル中のリードをリファレンスゲノムに対してマッピングを行う。マッピングするときに、インデックスを利用する。
このページでは、サンプルデータとして SRR1169893 と SRR1169894 を利用して、マッピングする例を示す。このデータはラットの RNA-seq データで、single-end リードである。
公共データベースからリファレンスゲノムをダウンロードする
ラットの全ゲノムデータは公共データベースである Ensembl でダウンローできる。しかし、全ゲノムをダウンロードしても、どの領域が遺伝子でどの領域がそうではないかのアノテーション情報が含まれていない。そのため、(マッピングには必要はないが、)全ゲノムの他にアノテーションデータも合わせてダウンロードすると便利である。
例えば、ラット関連のデータは Ensembl FTP で Rattus norvegicus の項でダウンロードできる。

上図の赤い枠で囲まれたファイルをダウンロードする。
- Ensembl FTP ページのラットの項目から、DNA(FASTA) 列にある FASTA ファイルをダウンロードする。その他に、cDNA、CDS や Protein sequence など様々な FASTA ファイルが用意されているが、ダウンロードするのはあくまで DNA(FASTA) の FASTA である。該当リンクをクリックすると、複数のファイルがリストアップされる。このうち、.toplevel.fa.gz で終わるファイルをダウンロードする(1 つだけあるはず)。このファイルが全ゲノム配列である。
※_sm.toplevel.fa.gz は利用しないので、ダウンロードする必要がない。 - 同様に、Ensembl FTP ページの Gene sets 列にある GTF リンクをクリックすると、アノテーションデータのダウンロードページに移動できる。.gtf.gz で終わるファイルをダウンロードする。(このファイルは、マッピング時に利用しないが、後々の解析がスムーズに進めるように予めダウンロードしておくとよい。)
これらは gzip 形式で圧縮されているため、ダウンロード後にこれらを展開する。
gunzip Rattus_norvegicus.Rnor_5.0.75.dna.toplevel.fa.gz
gunzip Rattus_norvegicus.Rnor_5.0.75.gtf.gz展開すると、リファレンスとなる全ゲノムファイル(Rattus_norvegicus.Rnor_5.0.75.dna.toplevel.fa)と アノテーションファイル (Rattus_norvegicus.Rnor_5.0.75.gtf)が得られる。
インデックスを作成する
リファレンスとなる全ゲノムのインデックスを作成する。このとき、bowtie2-build コマンドを利用する。-f の後に、ダウンロードした全ゲノムのファイル場所を指定し、その後にインデックスの名前を付ける。インデックス名前は任意で構わないが、ここでは RNOR75 とする。インデックスの作成は非常に時間がかかる。パソコンの性能によっては数時間以上及ぶ場合がある。
bowtie2-build -f Rattus_norvegicus.Rnor_5.0.75.dna.toplevel.fa RNOR75インデックスを作成し終えると、ディレクトリには 6 つのファイルが生成される。これら合わせて Bowtie2 のインデックスとなる。
- RNOR75.1.bt2
- RNOR75.2.bt2
- RNOR75.3.bt2
- RNOR75.4.bt2
- RNOR75.rev.1.bt2
- RNOR75.rev.2.bt2
拡張子が .bt2 のファイルが 6 つ生成される。ヒトゲノムのように、リファレンスが長い場合は拡張子が .bt2l になる。特に気にする必要はない。
Bowtie2 によるマッピング
Bowtie2 によるマッピングはリードとリファレンスのインデックスの 2 種類のデータを必要とする。
マッピングは以下のように bowtie2 コマンドを利用する。 サンプルデータは 2 つであるから、bowtie2 コマンドをそれぞれに対して実行する必要がある。
このサンプルデータは single-end リードであるから、-U のあとにその FASTQ ファイルを指定する。また、出力フォーマットを SAM 形式で出力する場合は -S を付ける。-q -N 1 -p 8 などは Bowtie2 のオプションを参考。
bowtie2 -q -N 1 -p 8 -x RNOR75 -U SRR1169893.fastq -S SRR1169893.sam
bowtie2 -q -N 1 -p 8 -x RNOR75 -U SRR1169894.fastq -S SRR1169894.samカウントデータの取得
次に HTSeq の htseq-count コマンドを利用してカウントデータの取得する。この際に、マッピング結果である SAM 形式のファイルと、アノテーションファイルである Rattus_norvegicus.Rnor_5.0.75.gtf を必要とする。
htseq-count SRR1169893.sam Rattus_norvegicus.Rnor_5.0.75.gtf > SRR1169893.counts.txt
htseq-count SRR1169894.sam Rattus_norvegicus.Rnor_5.0.75.gtf > SRR1169894.counts.txt実行が完了すると、SRR1169893.counts.txt などのファイルが生成される。これらのファイルの中にカウントデータが記載されている。
リファレンス上のどこか一箇所しかマッピングされないようなリードだけをカウントする場合は以下のように、XS を含む行を除いてカウントを行えば良い。以下の例では grep コマンドを利用して XS を含む行を除いている。
grep -v "XS:" SRR1169893.sam | htseq-count - Rattus_norvegicus.Rnor_5.0.75.gtf > SRR1169893.counts.txt
grep -v "XS:" SRR1169894.sam | htseq-count - Rattus_norvegicus.Rnor_5.0.75.gtf > SRR1169894.counts.txtカウントデータが複数のファイルにまたがっているため、これらを join コマンドで結合する。また、上述のカウントデータが記載されいてるファイルのなかに、遺伝子のカウントデータの他に、アノテーションがないリード数やマップされてなかったリード数なども保存されている。前者の場合は ENSG から始まり、後者の場合は「__no_feature」や「__not_aligned」などとなっている。そのため、grep 文を利用して ENSG だけから始まるデータのみを取得する。
# カウントデータのみを tmp.count.txt (一時ファイル)に保存する join -j 1 SRR1169893.count.txt SRR1169894.count.txt > tmp.count.txt # ヘッダーを all.count.txt に書き入れる echo "EnsemblID SRR1169893 SRR1169894" > all.count.txt # カウントデータを all.count.txt に書き入れる grep "^ENSG" tmp.count.txt >> all.count.txt # 一時ファイルを削除する rm tmp.count.txt
References
- Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012, 9(4):357-9. PubMed Abstract