生物の遺伝情報は細胞核の中にある DNA に記録されている。DNA の情報は、RNA ポリメラーゼにより転写され mRNA となり、核の外に運ばれる。核外に運ばれた mRNA は、リボソームによって翻訳されてタンパク質となり、様々な酵素活性に寄与している。DNA が mRNA に転写され、mRNA がさらにタンパク質に翻訳される一連の流れは、セントラルドグマと呼ばれている。
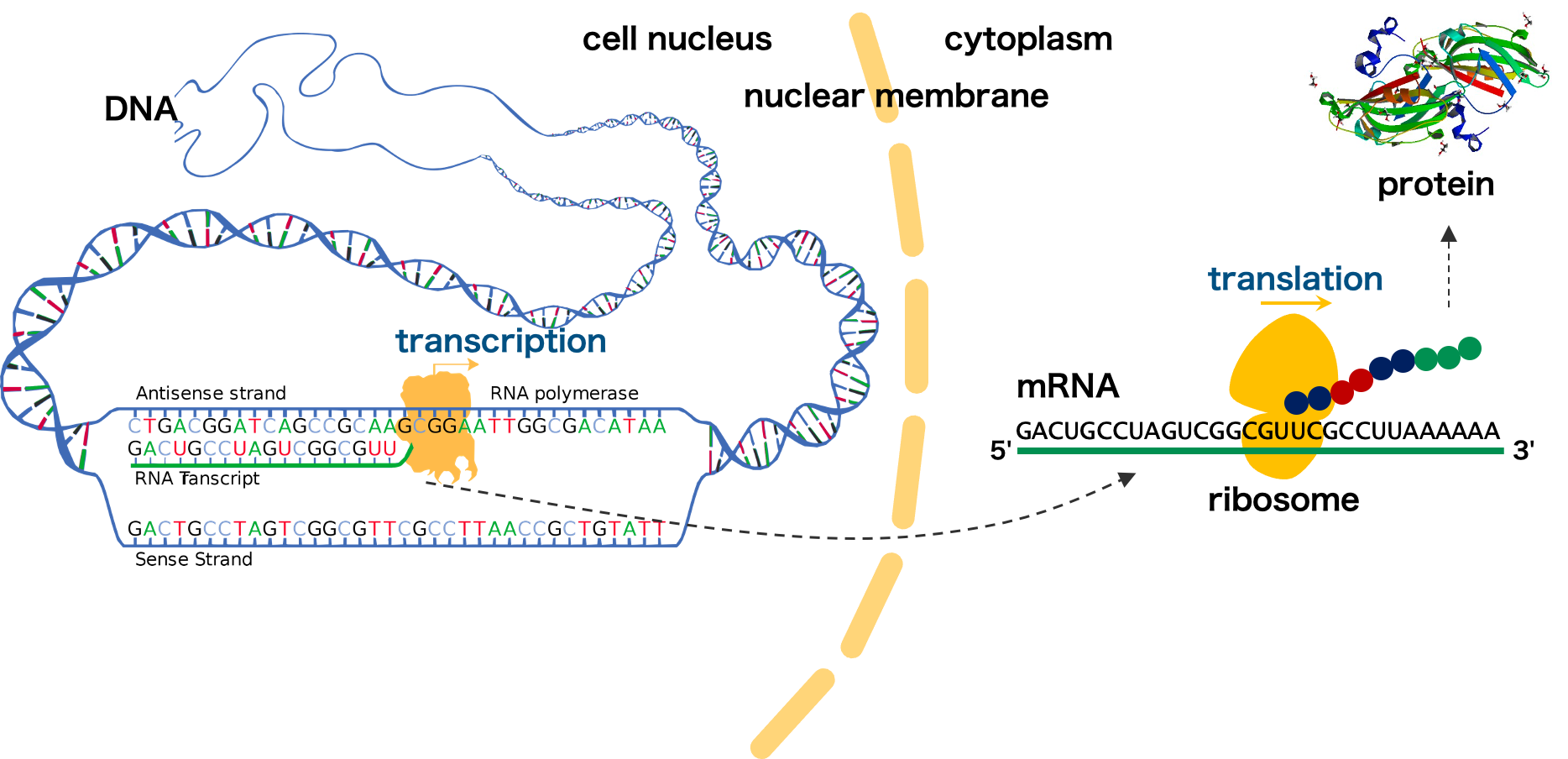
図は wikipedia より転載・変更を行った。
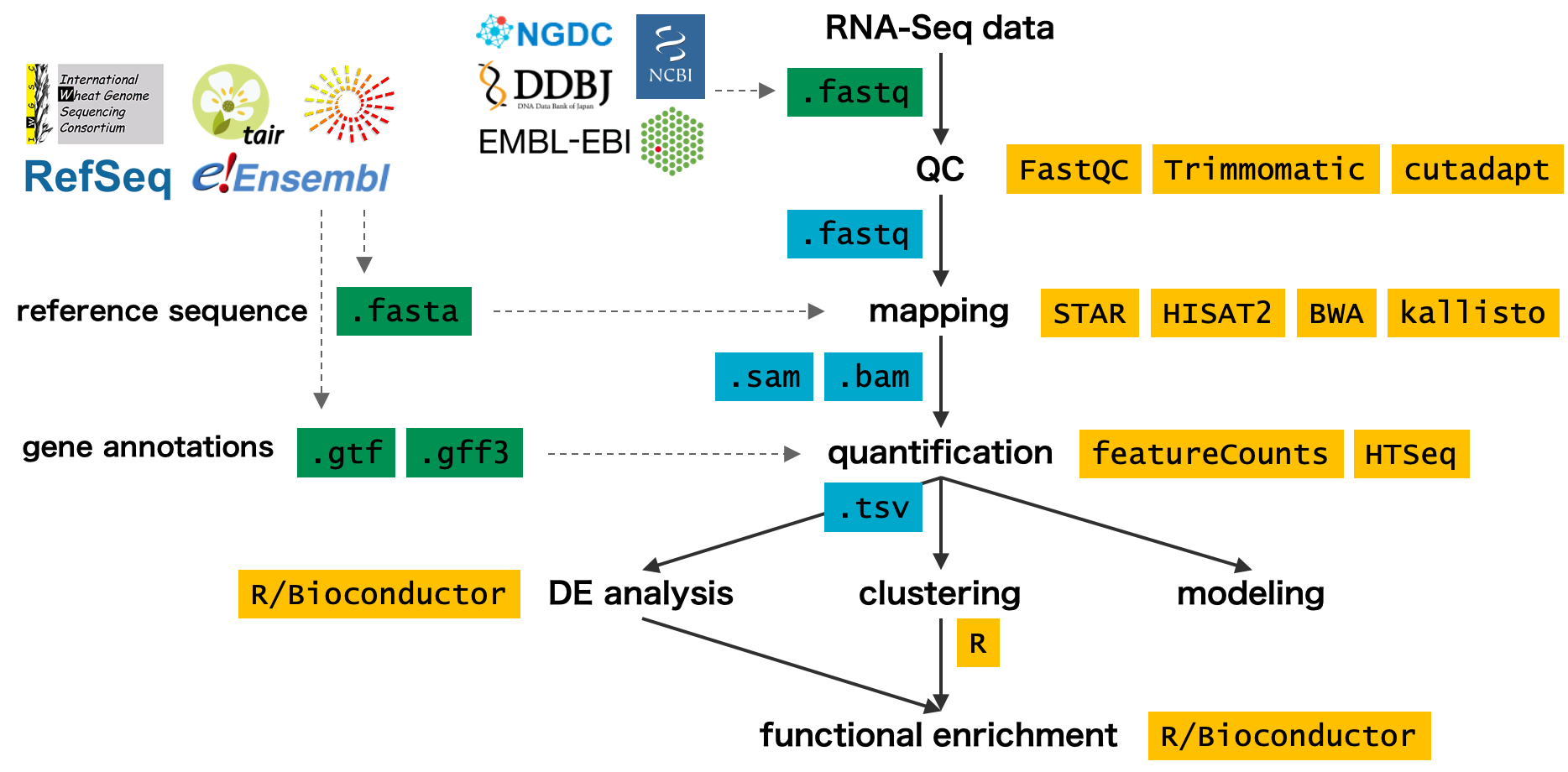
細胞の機能や生理活性は、細胞中の mRNA およびタンパク質に影響される。とくに、mRNA はタンパク質の生成にも寄与しているため、細胞の機能や生理活性を解明するための重要な因子となる。mRNA の量を調べる方法として、PCR、マイクロアレイや高速シーケンサーを利用した RNA-Seq がある。PCR による発現量定量は、信頼性が高いが、既知遺伝子の mRNA に対してしか適用できない。また、実験が煩雑で、一度に数個程度の mRNA をしか定量できない。これに対して、マイクロアレイおよび RNA-Seq は一度に数千から数万の遺伝子の mRNA を定量できる。特に、RNA-Seq では応用範囲が広く、配列が知られていない遺伝子の mRNA の定量も可能である。
RNA-Seq を利用した mRNA 定量では、細胞中の mRNA をすべて収集し、その塩基配列を高速シーケンサーで解読し、続けて、mRNA の塩基配列を見てそれがどの遺伝子に由来するのかを同定することで行われる。最後に、遺伝子ごとに何本の mRNA があったかを集計することで、その遺伝子の発現量を見積もることができるようになる。

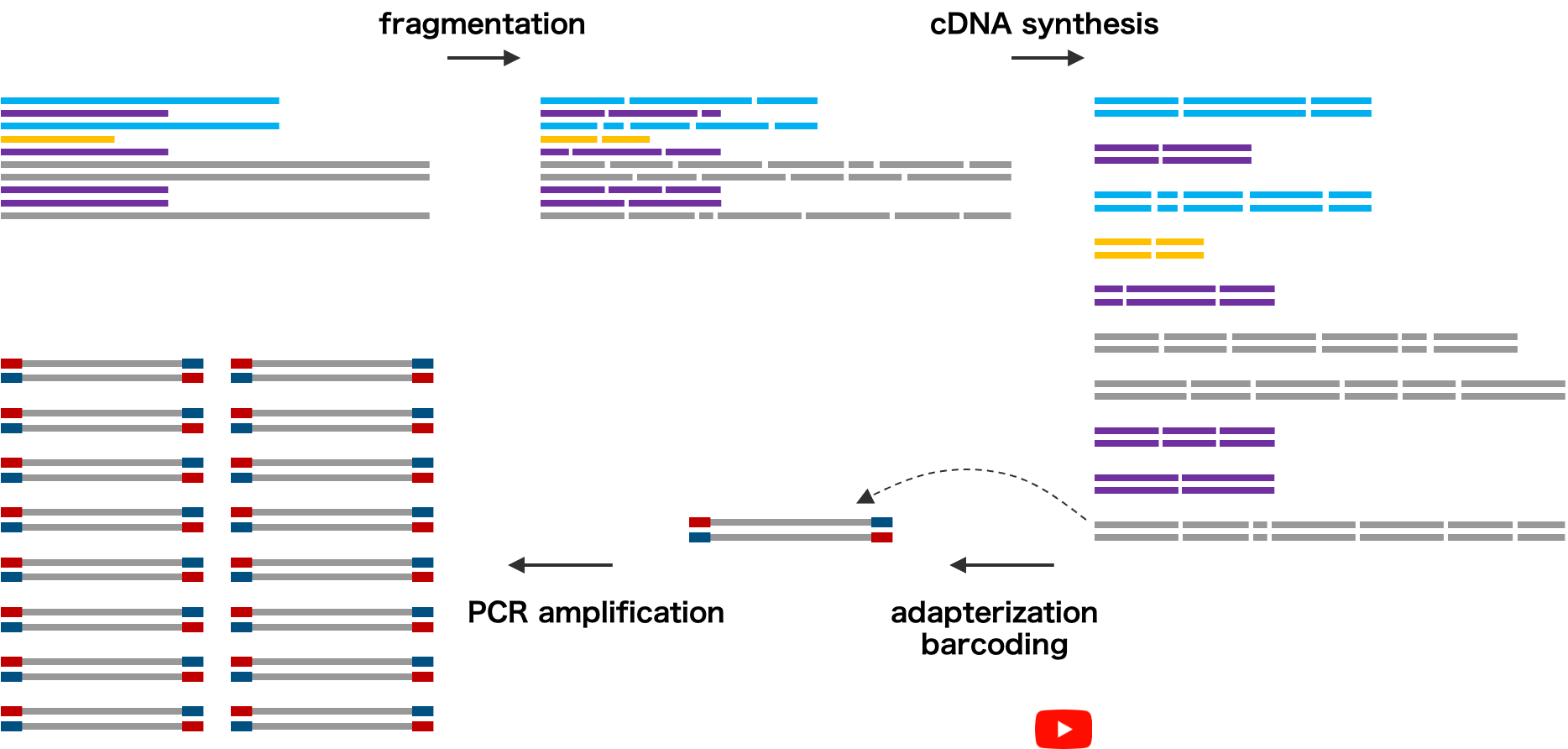
mRNA の長さは、短いものは 100 bp 前後であり、長いものは数キロに及ぶことが知られるている。生物種によって異なるが、mRNA の長さは概ね 500-2,000 bp の範囲に多く分布している。現在のシーケンス技術では、千キロ近くの塩基配列をシーケンスすることは可能だが、コストが非常に高い。そのため、現在では、長い mRNA をランダムに切断して、その短い断片の塩基配列をシーケンスする方法が取られている。シーケンスされたこれらの断片は、ジグゾーパズルのように繋ぎ直して解析を進める。
細胞中の mRNA を抽出してから高速シーケンサーにかけるまでのライブラリーの準備作業は、mRNA の断片化、cDNA の合成、バーコード配列・アダプター配列の取り付け、PCR 増幅の順で行う。mRNA は単鎖であるため分解されやすい。そのため、mRNA を断片化したのうち、すぐに cDNA の合成を行う必要がある。次に、cDNA の両端にアダプター配列やバーコード配列やアダプター配列を取り付けて、PCR 増幅を行う。PCR 増幅を行うことで、転写量の少ない遺伝子の mRNA 断片の量も指数的に増えて、高速シーケンサーによって検出されやすくなる。ただし、PCR 増幅の効率は塩基構成によって異なり、遺伝子の長さによっても異なることから、バイアスが生じる。そのため、現在では PCR 増幅を必要としない PCR-free の方法がスタンダードになりつつある。これらの作業は、それに適した実験キットが販売されている。実験キットに付属してる実験プロトコルに従ってライブラリー作成を行えばよく、必要に応じて、YouTube などの動画共有サイトの実験動画も参考になる。
調整したライブラリーを高速シーケンサーで処理すると、ライブラリー中にある mRNA 断片の塩基配列が読み取られる。その塩基配列データは FASTQ とよばれるフォーマットのテキストファイルに保存される。このファイルをバイオインフォマティックス解析することで、それぞれの断片がどの遺伝子の mRNA に由来するのかを同定することができ、最終的に各遺伝子(あるいは転写産物)の発現量を見積もることができるようになる。この解析は、以下のようにクオリティコントロール(QC)、マッピング、定量などからなる。