RNA-Seq 実験を行う上で、sequencing depth をどれぐらいにするのかが悩みのタネである。RNA-Seq を用いた遺伝子発現量解析が普及している今日でも、sequencing depth の決め方は、未だに標準となるものが存在しない。その理由として、sequencing depth は、着目している転写物の発現量や複雑さ、または解決しようとしている生物学的な問題を考慮して、case-by-case で決める必要があるから(Sims et al., 2014)。
sequencing depth とマッピングできたリードのバイアス
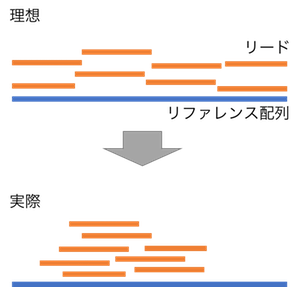
シーケンサーでシーケンシングされたリードは、必ずしも転写物のリファレンス配列に均一にマッピングされるわけではない。理想なケースとして、転写物がランダムに切断されて、その転写物断片がエラーなくシーケンシングされて、リードとなる。このようなリードを、転写物のリファレンス配列にマッピングすると、ほぼ均一にマッピングできる。しかし、実際ではこのようにならない。転写物の断片が切断される位置にやや特異性が見られる。例えば、Illumina のプロトコルを利用してライブラリー調整を行うと、シーケンシングされるリードの 5' 末端には特異性(5' 末端の 1 文字目は C である確率が高く、2 文字目は T である確率が高いなど)が見られる(Hansen et al., 2010)。このとき、シーケンシングされたリードは、リファレンス上の特定の箇所に多くマッピングされるようになる。
このように、リードはリファレンス配列に均一にマッピングされない。期待値よりも多くマッピングされた領域もあれば、まったくマッピングされない領域もある。このため、RNA-Seq 実験において、sequencing depth を決める際に、期待値よりも少なくマッピングされる領域もあることを考慮しなければならない。
sequencing depth と検出できる遺伝子数
1 つの細胞の中に存在する転写物は、1 コピーから数百万コピーと様々なレベルで存在している。コピー数の少ない転写物を検出したいのであれば、sequencing depth を増やせばよい。しかし、sequecing depth をむやみに増やしても、コストに見合う転写物を検出できるわけではない。
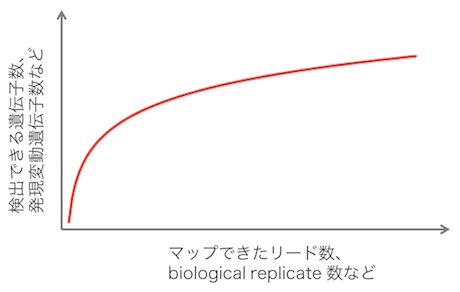
Sequencing depth が少ないとき、sequencing depth を増やすと、新たに検出できる転写物の数が劇的に増える。しかし、ある程度 sequencing depth が大きくなると、そこからいくら sequencing depth を増やしても、新たに検出できる転写物の数はほとんど増えなくなる。sequence depth と検出できる転写物数の関係は、生物種によって異なる。例えば、ヒトの場合は転写物の種類が多いためにより多くの sequencing depth を必要とするが、酵母の場合はより少ない sequencing depth で十分と考えられる。
ある生物種の sequence depth と検出される転写物数の関係を調べたいとき、希薄化曲線を利用すればよい。公共データベースで公開されているリードデータをダウンロードして、sequence depth を 1.0, 0.9, 0.8, 0.7, ... 倍にダウンサンプリングし、いくつかのサブセットを作成する。これらのサブセットに対して同一のマッピング処理を行い、検出できる転写物数を調べる。続いて、各サブセットの sequence depth と検出できる転写物数の対応を散布図にする。最後に、散布図上の各点にフィットするような近似曲線を描く。このような近似曲線(希薄化曲線)を描くことで、自分の目的にあった sequence depth の目安を決めることができるようになる。
sequencing depth と biological replicates
RNA-Seq 実験を行う上で、sequencing depth のほかに biological replicates の取り扱いも考慮しなければならない。限られた予算で、sequencing depth を増やすか、それとも biological replicates を増やすかが悩む問題である。sequencing depth を深くすることによって、発現量の低い転写物が検出できるようになる。しかし、sequencing depth を増やすために、biological replicate 無しにした場合、その低発現量の転写物は、その個体だけに見られるのか、それともその種一般にみられるのかという「生物学的なばらつき」が見積れなくなる。これに対して、biological replicate を多くした場合は、発現量の高い転写物しか検出できなくなる可能性があるが、解析結果がより再現性の高くなるというメリットがある。
References
- Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014, 15(2):121-32. DOI: 10.1038/nrg3642 PMID: 24434847
- Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010, 38(12):e131. DOI: 10.1093/nar/gkq224 PMID: 20395217
- Diminishing returns in next-generation sequencing (NGS) transcriptome data. Gene. 2015, 557(1):82-7. DOI: 10.1016/j.gene.2014.12.013 PMID: 25497830