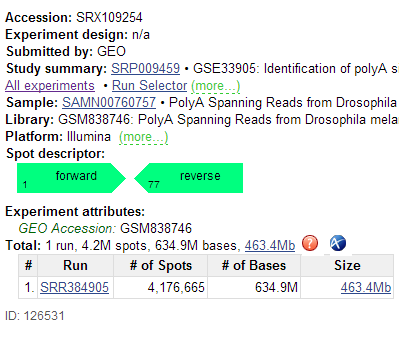
ペアエンドリードのデータのダウンロードは基本的にシングルエンドリードのダウンロード方法と同じ( 参照)。論文中にかかれている accession number を元に検索するが、直接 NCBI SRA のページにアクセスして accession number を調べる。ペアエンドリードの場合は、緑色のマークに forward と reverse がとなっているのを確認できる(NCBI SRA)。
表中の Size 項目の 463.4Mb のリンクをクリックすると、ダウンロードページに遷移する。青色のリンク(SRR384905.sra)をクリックすればダウンロードが開始される。
SRR384905 は SRP009459 プロジェクトのうちの 1 つのデータである。このプロジェクトはショウジョウバエの様々な組織のサンプルをシーケンスしたデータである。このプロジェクトの中のすべてのファイルをダウンロードする場合は、NCBI SRA のサイトにてすべてのデータをダウンロードする。全部で 58 ファイル(サンプル)ある。
上述の方法で 1 ファイルずつダウンロードも可能だが、ファイル数が多いときはシェルスクリプトを利用すると便利。ただし、あらかじめ、各ファイルのダウンロード URL の共通部分を調べておく必要がある。
# このプロジェクト中にある SRR 登録番号は 384905 から 384962 までであるから、以下のように登録番号からなる配列を作る。
sra_list=({384905..384962})
# ダウンロードする FTP サイトのパスを定義する。データによっては後半の SRP の部分が異なる。データに応じて事前にブラウザで調べる必要がある。
base_url=ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP009/SRP009459
# for 分を利用して 1 ファイルずつダウンロード
for sra_id in ${sra_list[@]}
do
wget ${base_url}/SRR${sra_id}/SRR${sra_id}.sra
sleep 10m
doneFASTQ への変換は、NCBI SRA toolkit の fastq-dump コマンドを利用する。この際に、ペアエンドリードを forward と reverse の 2 つのファイルに分けるために、 --split-files オプションを付ける。また、変換対象ファイルは ./ から書き始めないとエラーが起き正しく実行されない場合がある。
fastq-dump --split-files ./SRR384905.sra 実行が完了すると、RR384905_1.fastq と RR384905_2.fastq の 2 つのファイルができる。両者がそれぞれ forward と reverse に対応する。
シェルスクリプトでダウンロードしたすべての SRA ファイルを FASTQ に変換する場合は以下のようにする。
sra_list=({384905..384962})
for sra_id in ${sra_list[@]}
do
fastq-dump --split-files ./SRR${sra_id}.sra
done