遺伝子レベルのマッピング結果から得られたリードカウントデータから TPM あるいは FPKM を計算するとき、遺伝子の長さの定義が問題となる。遺伝子アノテーション(GFF や GTF)にある遺伝子の開始塩基の位置と終了塩基の位置の差をそのまま遺伝子の長さとした場合、その中にイントロンなどの非転写配列が含まれてしまい、正確に PFKM/TPM を計算できなくなる。また、1 遺伝子の遺伝子から複数のアイソフォームが生成されるので、アイソフォームによって特定のエクソンを使ったり、使わなかったりするため、すべてのエキソンの長さを足し合わせた長さを遺伝子長とした場合も、正確に FPKM/TPM を計算できない。このように、遺伝子の長さの定義は実に難しい問題である。
RSEM、Cufflinks、StringTie などのプログラムを使用すれば、各領域にマッピングされたリードのカバレッジを利用して、アイソフォームごとに TPM あるいは FPKM を正確に計算できる。一方で、解析者自身が、カウントデータから TPM または FPKM を計算する場合は、実験目的に応じて、次のように遺伝子長を定義して、計算することが多い。
- 遺伝子の転写産物のうち、もっとも長い転写産物の長さをその遺伝子の長さと定義する。
- 遺伝子の転写産物のうち、すべての転写産物の長さの平均値あるいは中央値をその遺伝子の長さと定義する。
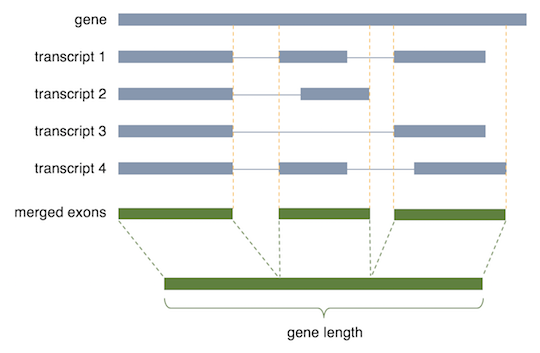
- 遺伝子領域上、エキソンになりうるすべての領域の長さをその遺伝子の長さと定義する。
3 番目のケースを図示すると次のようになる。
R
GFF または GTF ファイルをがあれば、R の GenomicFeature パッケージを利用すると、簡単に計算できる。
txdb <- makeTxDbFromGFF('Homo_sapiens.GRCh38.100.gtf', format = 'gtf')
exons_list_per_gene <- exonsBy(txdb, by = 'gene')
exonic_gene_sizes <- lapply(exons_list_per_gene, function(x){sum(width(reduce(x)))})
head(exonic_gene_sizes)
# $ENSG00000000003
# [1] 4536
# $ENSG00000000005
# [1] 1476
# $ENSG00000000419
# [1] 1207
# $ENSG00000000457
# [1] 6883
# $ENSG00000000460
# [1] 5970
# $ENSG00000000938
# [1] 3382Python
Python プログラムは次のようにかける。このプログラムの中身について、re.compile('gene_id "(ENSMUSG[0-9]+)";') のところで、文字列パターンマッチングにより遺伝子の名前を取得している。実際に使うとき、必要に応じて、このパターンを修正する必要がある。