正規化はデータ解析を行う上で欠かせない処理の一つである。重回帰分析を行うとき、説明変数として使う気温、降水量や日射量などのデータの単位が異なっていると、これらを標準化しなければならない。この標準化にあたる処理が、RNA-Seq データ解析において正規化とよばれている。RNA-Seq データの場合、各サンプルのデータはともにリードカウントであり、同じ単位である。したがって、単位を揃える処理を行う必要はない。実際に、RNA-Seq データの正規化では、主に総リード数を揃えたり、遺伝子長を揃えたりする処理になる。
RNA-Seq データ正規化で一般的に使われる方法は、解析目的によって大きく 2 種類に分けることができる。異なるサンプル間にある特定の遺伝子の発現量を比較するのが目的であれば、サンプル間の総リードカウントを補正する必要がある。これは、例えばストレス処理群と対照群にある各遺伝子に対して、その発現量を比較し、処理によって発現量が変化した発現変動遺伝子を検出する際によく利用される正規化方法である。具体例を挙げると、処理群の総リード数が 90 万リード、対照群の総リード数が 110 万リードであれば、両者を 100 万に揃えてから、処理群の遺伝子 a と対照群の遺伝子 a の発現量を比較して、発現量に差があるかどうかを検定する。解析対象のサンプルすべてに対して、総リード数を 100 万に揃える正規化法を CPM (count per million) と呼ぶ。CPM は、発現変動遺伝子を検出する前のクラスタリングや PCA を行うときに使われる。また、実際に発現変動遺伝子を検出するためには、CPM を少し補正した方法が使われている。このような正規化法は、edgeR に実装されている TMM 正規化 や DESeq/DESeq2 に実装されている RLE 正規化がよく知られている。サンプル間で同一遺伝子の発現量を比較するのが目的であれば、CPM、TMM、RLE で十分である。
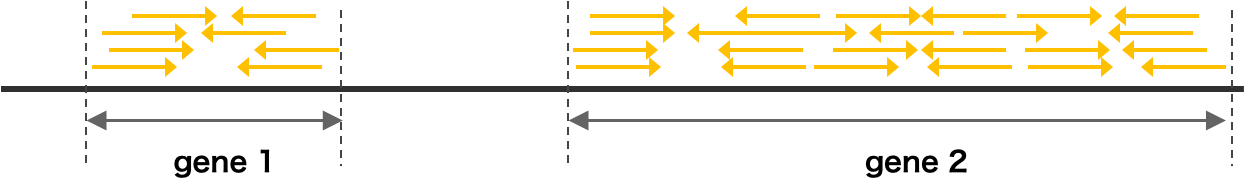
同じサンプルの中で複数の遺伝子の発現量を比較したい場合は、総リード数を揃えるだけの正規化法は不十分なことが多い。例えば、処理群にある遺伝子 1 と遺伝子 2 の発現量を比較したい場合は、処理群の総リード数を 100 万に揃えるだけでは意味がない。RNA-Seq でシークエンシングされるリードは、転写産物の断片の一部である。したがって、転写産物が長ければ、シークエンシングされる確率が高くなり、リード数が多くなる傾向がある。そのため、サンプル内の複数遺伝子の発現量を比較する場合は、総リード数を揃えるだけでなく、遺伝子長に対する補正も必要である。具体的に、リード数を転写産物(遺伝子)の長さが 1 kbp あたりのリード数に換算してから、総リード数を 100 万に揃える補正が行われる。このような正規化法として FPKM や TPM などが一般的に使われている。とくに近年では TPM を使用するのが一般的である。