MBCluster.Seq は RNA-seq のリードカウントデータの発現パターンに応じてクラスタリングを行うパッケージである。時系列あるいは組織別のプロファイルを得たいときに利用するとよい。
library(MBCluster.Seq)
packageVersion("MBCluster.Seq")
##[1] ‘1.0’サンプルデータ
サンプルデータは条件を設定して負の二項分布に従うように乱数生成により作成した。A 群と B 群の 2 つの実験群からなる。A 群には薬剤 A を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。B 群には薬剤 B を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。各実験の各時刻において 2 つの biological replicate があり、実験全体で計 12 biological replicate を使用したとする。また、全遺伝子数は 1,000 であり、その発現パターンは以下のように設定した。
| 遺伝子 | A 群の発現パターン | B 群の発現パターン |
| gene_1 ~ gene_40 | A6h > A3h > A0h, B6h > B3h > B0h A0h = B0h, A3h = B3h, A0h = B0h |
|
| gene_41 ~ gene_80 | A6h = A3h > A0h | A0h = B0h = B3h = B6h |
| gene_81 ~ gene_120 | A0h = A3h = A6h | A0h = B6h = B3h > B0h |
| gene_121 ~ gene_160 | A0h = A6h < A3h | A0h = B0h = B3h = B6h |
| gene_161 ~ gene_200 | A0h = A6h < A3h | A0h = B0h = B3h < B6h |
| gene_201 ~ gene_1000 | A0h = A3h = A6h = B0h = B3h = B6h | |
サンプルデータを読み込んでから行列型に変換し、count 変数に代入する。
count <- read.table("https://bi.biopapyrus.jp/data/counts_6_6_timecourse.txt", sep = "\t", header = T, row.names = 1)
count <- as.matrix(count)
dim(count)
## [1] 1000 12
head(count)
## A1_0h A2_0h A3_3h A4_3h A5_6h A6_6h B1_0h B2_0h B3_3h B4_3h B5_6h B6_6h
## gene_1 1 7 11 0 22 24 0 0 9 3 22 16
## gene_2 170 182 744 683 918 637 128 112 645 517 1248 1234
## gene_3 8 12 37 46 100 94 9 8 30 46 47 63
## gene_4 44 38 109 187 354 225 40 97 103 146 505 343
## gene_5 0 0 5 0 4 10 0 0 1 0 7 4
## gene_6 88 112 372 459 1010 611 129 80 486 360 678 767group にデータのラベルを記入する。group 変数については A 群と B 群の比較では group = c(rep(1, 6), rep(2, 6)) とするべきであるが、ここでは群間比較が目的ではなく、発現プロファイルのクラスタリングが目的であるから、A 群と B 群が独立しているほか、各時刻においてもそれぞれ独立したサンプルとして考えることができることから、
group を以下のように実験条件ごとに分けた。
group <- c(1, 1, 2, 2, 3, 3, 4, 4, 5, 5, 6, 6)クラスタリング
MBCluster.Seq を利用してクラスタリングを行うが、実際のクラスタリングを行う前に、MBCluster.Seq の関数を利用してデータを読み込んでおく必要がある。また、ここで利用するサンプルデータは上の表で示したように 6 パターンの発現プロファイルを持つことがわかっているので、クラスタサイズを 6 nK = 6 と指定してある。
dat <- RNASeq.Data(count, Normalizer = NULL,
Treatment = group, GeneID = rownames(count))
kmp <- KmeansPlus.RNASeq(data = dat, nK = 6)
cls <- Cluster.RNASeq(data = dat, model = "nbinom", centers = kmp$centers, method = "EM")クラスタリング結果は cls オブジェクトに保存されている。EM アルゴリズムを利用しているため、実行するたびに結果がやや異なってくることがある。
head(cls$probability)
## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 1.760936e-05 1.812046e-04 0.7869417 2.109541e-01 1.234020e-04 1.782049e-03
## [2,] 3.484958e-89 3.507966e-108 1.0000000 8.622396e-70 1.248952e-100 5.163926e-91
## [3,] 4.360068e-93 1.295790e-49 1.0000000 5.916831e-23 4.382738e-62 4.623404e-51
## [4,] 4.693138e-23 4.135958e-29 0.9999873 1.271856e-05 4.814205e-25 1.452876e-21
## [5,] 9.804749e-09 1.517993e-05 0.8923382 1.076324e-01 1.087917e-06 1.303337e-05
## [6,] 2.633411e-55 6.411368e-46 1.0000000 7.991553e-28 3.897313e-51 1.505876e-43
head(cls$cluster)
## [1] 3 3 3 3 3 3
table(cls$cluster)
## 1 2 3 4 5 6
## 51 96 51 54 72 676解析に用いたサンプルデータでは上の表に示したように、6 つの発現パターンがある。表の最後に示したパターンが 800 遺伝子であるのを除き、他のパターンはすべて 40 遺伝子となっている。解析結果を table 関数で見ると、クラスタ 6 は 676 遺伝子が含まれ、それ以外はのクラスタは 100 以下の遺伝子となっている。解析結果はやや妥当である。
次にこれを図示する。
tre <- Hybrid.Tree(data = dat, cluster0 = cls$cluster, model = "nbinom")
plotHybrid.Tree(merge = tre, cluster = cls$cluster, logFC = dat$logFC, colorful = TRUE)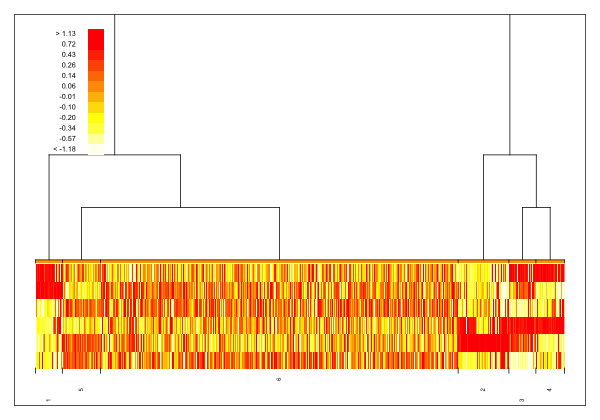
References
- Model-based clustering for RNA-seq data. Bioinformatics. 2014, 30(2):197-205. PubMed Abstract CRAN