maSigPro パッケージを利用して時系列データから発現変動遺伝子を検出する方法を示す。maSigPro はもともとマイクロアレイの時系列データ解析に使われているが、現在では次世代シーケンサーから得られるリードカウントデータも解析できるように拡張が行われた。
library(MASS)
library(maSigPro)
packageVersion("maSigPro")
##[1] ‘1.40.0’サンプルデータ
サンプルデータは条件を設定して負の二項分布に従うように乱数生成により作成した。A 群と B 群の 2 つの実験群からなる。A 群には薬剤 A を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。B 群には薬剤 B を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。各実験の各時刻において 2 つの biological replicate があり、実験全体で計 12 biological replicate を使用したとする。また、全遺伝子数は 1,000 であり、その発現パターンは以下のように設定した。
| 遺伝子 | A 群の発現パターン | B 群の発現パターン |
| gene_1 ~ gene_40 | A6h > A3h > A0h, B6h > B3h > B0h A0h = B0h, A3h = B3h, A0h = B0h |
|
| gene_41 ~ gene_80 | A6h = A3h > A0h | A0h = B0h = B3h = B6h |
| gene_81 ~ gene_120 | A0h = A3h = A6h | A0h = B6h = B3h > B0h |
| gene_121 ~ gene_160 | A0h = A6h < A3h | A0h = B0h = B3h = B6h |
| gene_161 ~ gene_200 | A0h = A6h < A3h | A0h = B0h = B3h < B6h |
| gene_201 ~ gene_1000 | A0h = A3h = A6h = B0h = B3h = B6h | |
サンプルデータを読み込んでから行列型に変換し、count 変数に代入する。
count <- read.table("https://bi.biopapyrus.jp/data/counts_6_6_timecourse.txt", sep = "\t", header = T, row.names = 1)
count <- as.matrix(count)
dim(count)
## [1] 1000 12
head(count)
## A1_0h A2_0h A3_3h A4_3h A5_6h A6_6h B1_0h B2_0h B3_3h B4_3h B5_6h B6_6h
## gene_1 1 7 11 0 22 24 0 0 9 3 22 16
## gene_2 170 182 744 683 918 637 128 112 645 517 1248 1234
## gene_3 8 12 37 46 100 94 9 8 30 46 47 63
## gene_4 44 38 109 187 354 225 40 97 103 146 505 343
## gene_5 0 0 5 0 4 10 0 0 1 0 7 4
## gene_6 88 112 372 459 1010 611 129 80 486 360 678 767maSigPro で解析する際に利用するデザインオブジェクトを作成する。
design <- data.frame(
Time = c(0, 0, 3, 3, 6, 6, 0, 0, 3, 3, 6, 6),
Replicates = c(1, 1, 2, 2, 3, 3, 4, 4, 5, 5, 6, 6),
Group.A = c(1, 1, 1, 1, 1, 1, 0, 0, 0, 0, 0, 0),
Group.B = c(0, 0, 0, 0, 0, 0, 1, 1, 1, 1, 1, 1)
)
rownames(design) <- colnames(count)発現変動遺伝子の検出
次に maSigPro を利用して、 A 群と B 群のデータを比較した際に、発現量に有意差をもつ遺伝子を検出していく。p.vector 関数を実行する前に必要であれば一度正規化してみるのもよい。また、発現量がすべてゼロの行が存在すると、p.vector 関数を実行するときに GLM 関連のエラーが生じるため、予め行合計がゼロの行を取り除きたほうがよい。
d <- make.design.matrix(design)
p <- p.vector(count, d, counts = TRUE)
t <- T.fit(p)解析結果は get.siggenes 関数を利用して取得する。
res <- get.siggenes(t, vars = "all")
head(res$sig.genes$sig.pvalues)
## p-value R-squared p.valor_beta0 p.valor_Group.BvsGroup.A
## gene_2 5.663454e-98 0.9846966 5.412354e-11 0.005281268
## gene_3 1.040187e-52 0.9690156 4.160167e-08 NA
## gene_4 1.041425e-12 0.8502937 4.857296e-10 NA
## gene_6 1.431419e-38 0.9521809 6.916261e-12 NA
## gene_7 5.231860e-11 0.7797428 3.536549e-03 NA
## gene_8 6.574223e-11 0.8450034 2.114619e-04 NA
## p.valor_Time p.valor_TimexGroup.B p.valor_Time2 p.valor_Time2xGroup.B
## gene_2 3.930817e-06 NA 7.612456e-05 0.001298054
## gene_3 5.735014e-05 NA 1.363964e-02 0.008595316
## gene_4 2.801832e-05 NA NA NA
## gene_6 6.014336e-05 NA 9.429070e-03 NA
## gene_7 1.043090e-04 0.03488876 NA NA
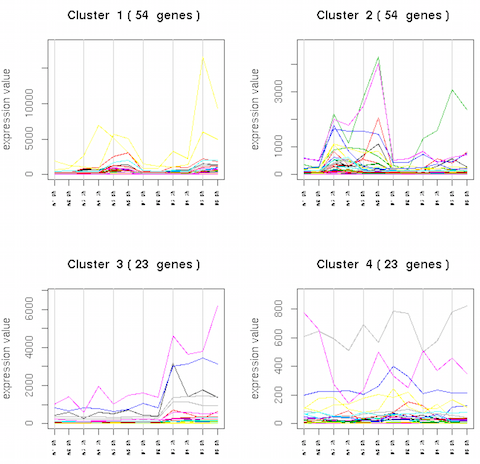
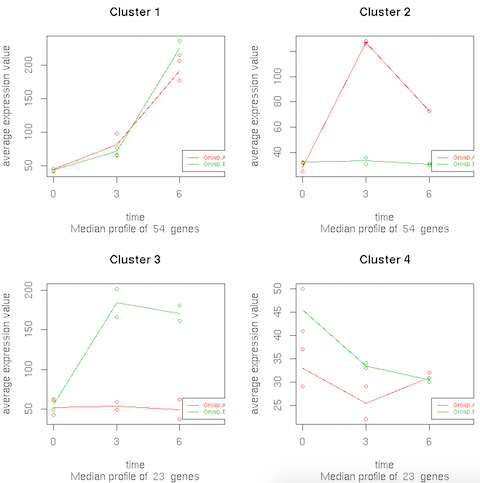
## gene_8 9.874559e-04 NA 1.118752e-02 NA解析に用いたサンプルデータには 4 パターンの発現変動遺伝子が存在することがわかっているので、ここでは、解析結果を 4 つのクラスタに分けてみてみる。gene_1 〜 gene_40 は群間に差がないので、群間比較の際に発現変動遺伝子といえないが、なぜかそのパターンが検出されている。
cl <- see.genes(res$sig.genes, k = 4) head(cl$cut) ## gene_2 gene_3 gene_4 gene_6 gene_7 gene_8 ## 1 1 1 1 1 1 tail(cl$cut) ## gene_848 gene_897 gene_936 gene_942 gene_954 gene_999 ## 4 1 3 3 2 4
References
- maSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics. 2006, 22(9):1096-102. PubMed Abstract Bioconductor
- maSigPro: Significant Gene Expression Profile Differences in Time Course Microarray Data. Website