DESeq2 は RNA-seq のリードカウントデータから発現変動遺伝子を検出するためのパッケージである。一般化線形モデルをサポートしているため複雑な解析にも柔軟に対応できる。二群間比較に対しては、Wald 検定と尤度検定の 2 種類の方法が利用できる。
R を起動して DESeq2 を呼び出す。呼び出しが完了したら DESeq2 のバージョンをチェックする。このページではバージョン 1.8.0 の解析結果を示している。バージョンが異なると解析結果が異なる場合がある。
library(DESeq2)
packageVersion("DESeq2")
##[1] ‘1.8.0’サンプルデータ
サンプルデータは、カウントデータが負の二項分布に従うように乱数を生成して作成した。A 群と B 群の 2 群からなり、それぞれの実験群には 3 つの biological replicate が含まれる。また、全遺伝子数は 1,000 個であり、その発現パターンは以下のように設定した。
| gene_1 ~ gene_160 | B 群に比べ、A 群での発現量が高い |
| gene_161 ~ gene_200 | A 群に比べ、B 群で発現量が高い |
| gene_201 ~ gene_1000 | 両群において差異なし |
サンプルデータを読み込んでから行列型に変換し count 変数に代入する。
count <- read.table("https://bi.biopapyrus.jp/data/counts_3_3.txt",
sep = "\t", header = T, row.names = 1)
count <- as.matrix(count)
dim(count)
## [1] 1000 6
head(count)
## A1 A2 A3 B1 B2 B3
## gene_1 3 2 40 4 2 0
## gene_2 680 807 594 165 91 73
## gene_3 35 38 31 7 9 11
## gene_4 103 165 110 38 31 28
## gene_5 2 9 1 3 1 0
## gene_6 419 373 399 133 89 58実験群の識別ラベルを作成し、group 変数に代入する。
group <- data.frame(con = factor(c("A", "A", "A", "B", "B", "B")))Wald 検定
DESeq2 を利用して発現変動遺伝子を検出する簡単な例として、以下のように 3 行で解析結果が得られる。1 行目でリードカウントと実験群のラベルを一つの変数(オブジェクト)に格納する。2 行目で正規化、分散の推測および検定を行う。3 行目で検定結果を整理して出力させる。
dds <- DESeqDataSetFromMatrix(countData = count, colData = group, design = ~ con)
dds <- DESeq(dds)
res <- results(dds)
head(res)
## log2 fold change (MAP): con B vs A
## Wald test p-value: con B vs A
## DataFrame with 6 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## gene_1 8.059985 -1.5684609 0.8904756 -1.7613745 NA NA
## gene_2 383.730021 -2.4023514 0.3746808 -6.4117280 1.438794e-10 4.370336e-09
## gene_3 20.998313 -1.6414727 0.5120116 -3.2059288 1.346273e-03 9.621893e-03
## gene_4 76.197165 -1.7331538 0.3823303 -4.5331323 5.811539e-06 6.140017e-05
## gene_5 2.589502 -0.7404315 0.9041106 -0.8189612 4.128085e-01 NA
## gene_6 235.296812 -1.8614974 0.3602019 -5.1679283 2.367031e-07 3.521563e-06
## head(res)検定結果を MA プロットに描く際に plotMA 関数を利用する。この際に、p-value が 0.01 よりも小さい遺伝子を赤くする。
plotMA(res, alpha = 0.01)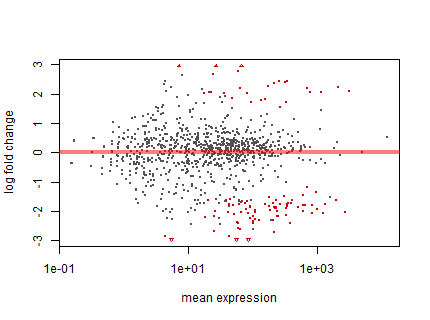
DESeq 関数は内部的に正規化、分散の推測および検定を行う関数を実行している。そのため、解析の自由度を高めるために、DESeq 関数を利用せずに、以下のように行ってもよい。
dds <- DESeqDataSetFromMatrix(countData = count, colData = group, design = ~ con)
dds <- estimateSizeFactors(dds)
dds <- estimateDispersions(dds)
dds <- nbinomWaldTest(dds)
res <- results(dds)
head(res)
## log2 fold change (MAP): con B vs A
## Wald test p-value: con B vs A
## DataFrame with 6 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## gene_1 8.059985 -1.5684609 0.8904756 -1.7613745 NA NA
## gene_2 383.730021 -2.4023514 0.3746808 -6.4117280 1.438794e-10 4.370336e-09
## gene_3 20.998313 -1.6414727 0.5120116 -3.2059288 1.346273e-03 9.621893e-03
## gene_4 76.197165 -1.7331538 0.3823303 -4.5331323 5.811539e-06 6.140017e-05
## gene_5 2.589502 -0.7404315 0.9041106 -0.8189612 4.128085e-01 NA
## gene_6 235.296812 -1.8614974 0.3602019 -5.1679283 2.367031e-07 3.521563e-06解析結果を result.txt ファイルに保存する。
write.table(res, file = "result.txt", row.names = T, col.names = T, sep = "\t")尤度比検定
尤度比検定は一般化線形モデルの検定でよく使われる検定の一つである。検定を行うには 2 つのモデルを用意する必要がある。それぞれモデルから尤度を計算し、比較する。尤度の比較を行うことにより、2 つのモデルが同じかどうかを検定する。例えば、モデル F は β1 と β2 の 2 つのパラメーターから構成されたものとする。また、モデル R は β1 だけから構成されたものとする。ここで、モデル F とモデル R の尤度を比較し、もし両者の尤度に有意差が見られるならば、2 つのモデルが異なるモデルであることを意味する。言い換えれば、β2 の効果により、モデル F とモデル R に差異をもたらした。すなわち β2 ≠ 0 と考えられる。逆にもし両者の尤度に有意差が見られなければ、β2 の効果が認められないこととなり、β2 = 0 と考えられる。
いま、遺伝子 i に着目して尤度比検定を説明する。遺伝子 i の A 群でのカウントデータを YiA とし、その期待値は E[YiA] と表せる。同様に、B 群でのカウントデータの YiB とし、その期待値は E[YiB] と表せる。ここでパラメーター βi1 を仮定し、E[YiA] = βi1 とする。次に、もう 1 つのパラメーター βi2 を仮定し、E[YiB] = βi1 + βi2 とする。これを行列式で記述すると以下のようになる。
この仮定で検定について考える。検定を考えるとはまず帰無仮説を立てなければならない。ここでは A 群の発現量と B 群の発現量に差異があるかどうかを検定するのが目的である。このとき帰無仮説を「A 群の発現量と B 群の発現量に差異がない」とすることができる。すると、これは「βi2 = 0」に等しい。βi2 = 0 のときの各群の期待値は以下のように記述できる。
ここまで行列で記述した E[Y] = Xβ の形をした数式をモデルという。1 つ目のモデルは想定されたすべてのパラメーターが使われていることから full model と呼ばれる。2 つ目のモデルは full model からパラメーターが 1 つ取り除かれて(βi2 = 0)いることから reduced model と呼ばれる。また、行列 X をデザイン行列または計画行列という。
次に full model と reduced model のそれぞれから尤度を計算する。両者の尤度を比較する。もし両者の尤度に有意差が認められれば、これは full model と reduced model が相違異なるモデルであることを意味する。このとき帰無仮説を棄却することができ、すなわち、βi2 ≠ 0 あるいは、E[YiA] ≠ E[YB] と判定を下すことができる。逆に、両者の尤度に有意差が認められなければ、full model と reduced model が同じと考えられて帰無仮説は保留される、すなわち βi2 = 0 すなわち E[YiA] ≠ E[YB] の判定を下せなくなる。このような作業を遺伝子 1 から遺伝子 n まで繰り返していく。
(仮説検定においてここで注意したいたのは、帰無仮説が保留される(棄却されない)とき、E[YiA] = E[YB] を意味しているわけではない、あくまで判定不可能であり、判断を保留することである。)
このように方法で検定を行う方法を尤度比検定という。上述では E[Y] = Xβ と記述したが、正しくは g(E[Y]) = Xβ と記述すべきである。関数 g はリンク関数と呼ばれている。正規分布では g(x) = x であり、リードカウントデータが従う負の二項分布では g(x) = log(x) が使われている。データの従う分布が決まれば、リンク関数の形もほぼ決められているため、R で解析を行う際にリンク関数のことを考慮せずに、右辺の Xβ だけに着目すればよい。このページおいては、説明の簡潔のためにリンク関数を省略して書く。
では、実際にサンプルデータをモデル化して edgeR で解析していく。ある遺伝子 1 つだけに着目したとき、A 群の発現量を β1 とおく。次に、A 群の発現量と B 群の発現量の差を β2 とおく。このとき、E[A] = β1、E[B] = β1 + β2 である。サンプルデータは 3 × 3 の biological replicate からなるから、モデルは以下のようになる。
遺伝子が n 個があればこのような式は n 個できる。遺伝子が n 個あるときプログラムが逐次的に処理を行ってくれるため、遺伝子が 1 個だけの時を考えればよい。
A 群と B 群の発現量を比較したいので、帰無仮説として A 群と B 群の発現量は同じであると考えればよい。そこで帰無仮説を検定するために、1 つ目のモデルと比較するためのモデルを以下のように作成できる。
ここまで作成した 2 つのモデルはそれぞれ full model と reduced model という。前者は想定されるパラメーター β1 と β2 をすべて含んでいることから full model と呼ばれている。後者は full model からパラメーター β2 を取り除いてできたことから reduced model と呼ばれる。
full model のデザイン行列 model.matrix 関数を利用して簡単に作成できる。
model.matrix( ~ group$con)
## (Intercept) group$conB
## 1 1 0
## 2 1 0
## 3 1 0
## 4 1 1
## 5 1 1
## 6 1 1
## attr(,"assign")
## [1] 0 1
## attr(,"contrasts")
## vattr(,"contrasts")$`group$con`
## [1] "contr.treatment"一方、reduced model のデザイン行列につていは作成できないので、1 列目だけを 1 、他の列を 0 にするという意味で ~ 1で代用する。
ここで、DESeq2 を利用して尤度比検定を行う。nbinomLRT 関数を利用するとき、引数 full には full model を作成する際に使用した式、引数 reduced には reduced model を作成する際に使用した式を代入する。DESeqDataSetFromMatrix 関数を利用するときに colData 引数に group 変数が与えられているため、nbinomLRT では group$con のように入力する代わりに、省略して con を入力する。
dds <- DESeqDataSetFromMatrix(countData = count, colData = group, design = ~ con)
dds <- estimateSizeFactors(dds)
dds <- estimateDispersions(dds)
dds <- nbinomLRT(dds, full = ~ con, reduced = ~ 1)
res <- results(dds)
head(res)
## log2 fold change (MLE): con B vs A
## LRT p-value: '~ con' vs '~ 1'
## DataFrame with 6 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## gene_1 8.059985 -2.752421 1.5523197 2.9133749 8.784787e-02 3.509140e-01
## gene_2 383.730021 -2.515429 0.3925618 37.2077335 1.061918e-09 2.787535e-08
## gene_3 20.998313 -1.800921 0.5647963 10.1300056 1.458750e-03 1.051158e-02
## gene_4 76.197165 -1.818558 0.4017903 19.8395419 8.422225e-06 9.103435e-05
## gene_5 2.589502 -1.445497 1.7622123 0.6641555 4.150967e-01 NA
## gene_6 235.296812 -1.941838 0.3759501 25.1870331 5.203086e-07 7.648536e-06解析結果を result.txt ファイルに保存する。
write.table(res, file = "result.txt", row.names = T, col.names = T, sep = "\t")二群間比較(biological replicate なし)
DESeq2 は、biological replicate なしの二群間比較に対応していない。biological replicate なしの二群間比較用のデータを与えると、検定を行なって、p-value などを出力するが、発現量が大きくかつ fold-change が大きくても、p-value が大きかったりするような結果が得られる。DESeq2 を利用して、biological replicate なしの二群間比較を行う場合は、検定結果を MA プロットに描き出して、一度確認した方がいいかもしれない。
References
- Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014. 15(12):550. PubMed Abstract
- Differential expression analysis for sequence count data. Genome Biol. 2010, 11(10):R106. PubMed Abstract
- 一般化線形モデル. biopapyrus バイオスタティスティックス