RNA-Seq を用いた解析で、多数のサンプル(またはライブラリー)を対象としているとき、サンプル同士の類似度を調べたい場合がある。類似度を調べる方法としては階層クラスタリング、主成分分析(PCA)、独立成分分析(ICA)や非負行列分解(NMF)などの方法が挙げられる。ここでは、NMF について紹介する。NMF のアルゴリズムの特徴として非負数の行列を入力として、これを 2 つの非負数の行列に分解するというものである。RNA-Seq から得られる遺伝子発現量は 0 から数千・数万までの値をとることから、遺伝子発現量行列がうまく分解されない場合が多い。
非負行列分解アルゴリズム
非負行列分解(Non-negative Matrix Factorization)は、ある 1 つの行列を、2 つの小さな行列に分解することである。ここで、ある行列を N×M の行列 A とし、行列 A が行列 W および行列 H の積として表せるものとする。
なお、行列 W は N×k、行列 H は k×M の行列であり、また、 k をランクと呼び、 k ≤ min{N, M} を満たす。行列分解を行う際に、一般的にランク k を k << min{N, M} を満たすように選ぶ。
一般的な解き方としては ||A - WH||2 が最小となるように、H と W を求めていく。まず、行列 W と行列 H をランダムなど非負数の値で埋める。次に、H を固定して W を最適化してから、今度は W を固定して H を最適化していく。これを繰り返しながら 2 つの行列を計算していくというものである。R では nmf または NMF パッケージにこのような機能が実装されている。
サンプルデータ
ここで乱数の生成により作成したデータを利用して非負行列分解を行ってみる。サンプルデータは条件を設定して負の二項分布に従うように乱数生成により作成した。A 群と B 群の 2 つの実験群からなる。A 群には薬剤 A を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。B 群には薬剤 B を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。各実験の各時刻において 2 つの biological replicate があり、実験全体で計 12 biological replicate を使用したとする。また、全遺伝子数を 1,000 とした。
サンプルデータを読み込んでから行列型に変換し、count 変数に代入する。
count <- read.table("https://bi.biopapyrus.jp/data/counts_6_6_timecourse.txt", sep = "\t", header = T, row.names = 1)
count <- as.matrix(count)
dim(count)
## [1] 1000 12
head(count)
## A1_0h A2_0h A3_3h A4_3h A5_6h A6_6h B1_0h B2_0h B3_3h B4_3h B5_6h B6_6h
## gene_1 1 7 11 0 22 24 0 0 9 3 22 16
## gene_2 170 182 744 683 918 637 128 112 645 517 1248 1234
## gene_3 8 12 37 46 100 94 9 8 30 46 47 63
## gene_4 44 38 109 187 354 225 40 97 103 146 505 343
## gene_5 0 0 5 0 4 10 0 0 1 0 7 4
## gene_6 88 112 372 459 1010 611 129 80 486 360 678 767ここで解析目的に応じて、発現量をあらかじめ正規化したり、発現量の小さい遺伝子を取り除いたりして、対数化したりしてから次の解析に進む。
count.log <- log10(count + 1)R による非負行列分解
カウントデータを一度対数化してから非負行列分解する。また、ここでは rank をライブラリー数(12 ライブラリー)に設定しているが、このサンプルデータは 2 群間 × 3 時点のデータであるので、rank を 6 に設定してもよい(解釈しやすい)。一般的にランクは、行数あるいは列数の小さい方の数値よりも更に小さい値を選ぶ。
library(NMF)
res <- nmf(count.log, rank = ncol(count.log), seed = 123456)非負行列分解の結果が res に保存される。非負行列分解により得られた行列 W および行列 H はそれぞれ basis および coef で確認できる。
w <- basis(res)
head(w)
## [,1] [,2] [,3] [,4] [,5] [,6]
## gene_1 4.867187 4.598508e+00 5.800671e-01 2.220446e-16 2.220446e-16 2.124390
## gene_2 5.427483 4.365396e+00 4.263003e+00 2.455375e+00 4.861098e+00 4.619308
## gene_3 4.048369 3.858420e+00 1.813159e+00 1.597343e+00 9.380248e-03 3.125106
## gene_4 3.707217 4.230725e+00 3.155016e+00 1.872344e+00 1.842737e+00 3.975692
## gene_5 3.276444 2.220446e-16 2.220446e-16 2.220446e-16 2.220446e-16 1.091318
## gene_6 5.375132 4.485551e+00 3.707988e+00 3.008995e+00 3.697905e+00 4.684141
## [,7] [,8] [,9] [,10] [,11]
## gene_1 1.498835e+00 2.816944e-07 1.187040 6.727647e-07 2.220446e-16
## gene_2 4.742552e+00 4.992536e+00 5.526517 3.863678e+00 5.926318e+00
## gene_3 2.974296e+00 2.203345e+00 2.268389 1.729149e+00 3.736408e+00
## gene_4 3.822356e+00 4.458201e+00 3.819983 2.870276e+00 4.818748e+00
## gene_5 2.220446e-16 1.804020e+00 2.067091 2.220446e-16 2.220446e-16
## gene_6 4.189414e+00 4.262592e+00 4.806549 3.610408e+00 5.571934e+00
## [,12]
## gene_1 2.220446e-16
## gene_2 3.610850e+00
## gene_3 2.317348e+00
## gene_4 4.407443e+00
## gene_5 2.220446e-16
## gene_6 3.613967e+00
h <- coef(res)
head(h)
## A1_0h A2_0h A3_3h A4_3h A5_6h
## [1,] 1.218325e-15 2.220446e-16 6.702316e-02 1.035056e-10 1.921045e-14
## [2,] 1.551982e-04 2.850231e-01 1.987015e-02 1.901340e-06 7.329117e-05
## [3,] 5.190363e-01 8.649945e-14 2.240581e-03 5.056638e-11 7.985852e-13
## [4,] 8.017608e-03 8.407642e-08 3.078928e-12 2.498992e-07 4.477024e-09
## [5,] 1.439755e-10 2.100516e-01 1.107513e-01 1.307652e-12 3.677396e-13
## [6,] 4.665122e-10 1.457697e-11 3.156218e-04 1.641273e-09 6.410054e-01
## A6_6h B1_0h B2_0h B3_3h B4_3h
## [1,] 3.663993e-01 2.692976e-06 3.663324e-13 1.171800e-01 2.220446e-16
## [2,] 2.331701e-03 2.256314e-10 4.192188e-12 8.283877e-13 5.356390e-03
## [3,] 2.118996e-10 6.518979e-12 1.379771e-10 6.657247e-07 3.530437e-16
## [4,] 2.137934e-11 3.721552e-01 1.524238e-09 9.995744e-02 1.836155e-01
## [5,] 2.220446e-16 6.101351e-02 1.192808e-01 1.129466e-05 3.670926e-14
## [6,] 1.043611e-08 2.040745e-07 5.063707e-12 1.461154e-11 8.805548e-12
## B5_6h B6_6h
## [1,] 6.716198e-13 2.399810e-16
## [2,] 1.651000e-01 2.516469e-01
## [3,] 1.517949e-04 3.328082e-14
## [4,] 7.205508e-16 1.704080e-11
## [5,] 2.220446e-16 2.759260e-15
## [6,] 1.055000e-11 1.678104e-10また、非負行列分解により計算された行列 W および行列 H の積は fitted(res) または w %*% h で確認できる。分解前のデータに近いことがわかる。コンピューターで扱える少数の桁数は有限であるので、分解前と一致する結果が得られない。
head(fitted(res))
## A1_0h A2_0h A3_3h A4_3h A5_6h A6_6h
## gene_1 3.017896e-01 1.310681e+00 0.7058264 8.828855e-06 1.3620823 1.794056
## gene_2 2.233029e+00 2.265327e+00 2.8588630 2.839882e+00 2.9633229 2.808495
## gene_3 9.545069e-01 1.101712e+00 1.4807035 1.714320e+00 2.0043885 2.011954
## gene_4 1.653246e+00 1.592929e+00 2.0272443 2.279917e+00 2.5502372 2.356503
## gene_5 5.092440e-10 1.590981e-11 0.2927942 2.150438e-09 0.6995407 1.200487
## gene_6 1.949413e+00 2.055243e+00 2.5588409 2.667670e+00 3.0047591 2.790293
## B1_0h B2_0h B3_3h B4_3h B5_6h B6_6h
## gene_1 1.375251e-05 0.0166616479 0.7207108 7.184682e-01 0.7593027 1.547604
## gene_2 2.110710e+00 2.0520918915 2.8075979 2.718819e+00 3.0956747 3.093443
## gene_3 9.923803e-01 0.9435804058 1.4701056 1.712828e+00 1.6899651 1.830791
## gene_4 1.613207e+00 1.9911134278 2.0162960 2.172424e+00 2.7027083 2.537408
## gene_5 3.253336e-01 0.0002387076 0.6457912 9.631020e-12 0.5775766 0.679842
## gene_6 2.114138e+00 1.9078149564 2.6852157 2.561834e+00 2.8308678 2.887044
head(count.log)
## A1_0h A2_0h A3_3h A4_3h A5_6h A6_6h B1_0h
## gene_1 0.3010300 0.903090 1.0791812 0.000000 1.361728 1.397940 0.000000
## gene_2 2.2329961 2.262451 2.8721563 2.835056 2.963316 2.804821 2.110590
## gene_3 0.9542425 1.113943 1.5797836 1.672098 2.004321 1.977724 1.000000
## gene_4 1.6532125 1.591065 2.0413927 2.274158 2.550228 2.354108 1.612784
## gene_5 0.0000000 0.000000 0.7781513 0.000000 0.698970 1.041393 0.000000
## gene_6 1.9493900 2.053078 2.5717088 2.662758 3.004751 2.786751 2.113943
## B2_0h B3_3h B4_3h B5_6h B6_6h
## gene_1 0.0000000 1.000000 0.602060 1.361728 1.230449
## gene_2 2.0530784 2.810233 2.714330 3.096562 3.091667
## gene_3 0.9542425 1.491362 1.672098 1.681241 1.806180
## gene_4 1.9912261 2.017033 2.167317 2.704151 2.536558
## gene_5 0.0000000 0.301030 0.000000 0.903090 0.698970
## gene_6 1.9084850 2.687529 2.557507 2.831870 2.885361非負行列分解の結果の図示
非負行列分解は ||A - WH||2 が最小となるように繰り返し計算を行っている。繰り返し毎にどれぐらいの残差になるかを図示する場合、nmf 関数を利用するとき .options 引数を与えれば良い。
res <- nmf(count.log, rank = ncol(count.log), seed = 123456, .options = "t")
plot(res)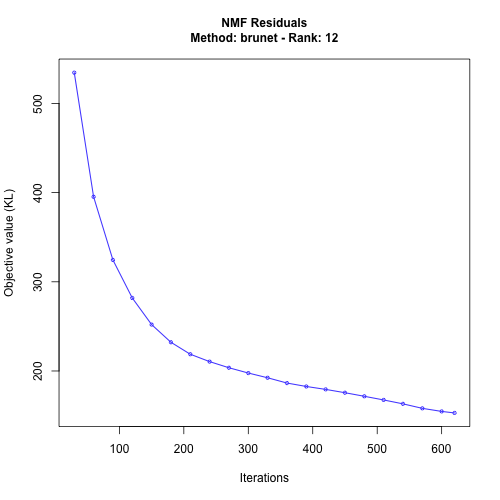
行列 W および行列 H のヒートマップはそれぞれ basismap および coefmap 関数で描くことができる。
layout(cbind(1, 2))
basismap(res)
coefmap(res)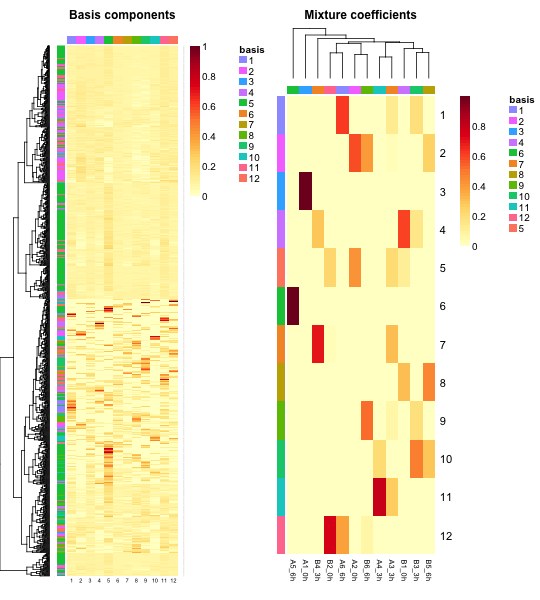
非負行列分解のランクについて
次元削減のために、ライブラリー数よりも少ない列数(ランク)で行列分解を行うこともある。しかし、どのランクがもっともよいのかはいろいろと検討する必要がある。そこで NMF を利用するとき、rank に複数の値を与えて計算したあとに、適切さを確認することができる。
res <- nmf(count.log, rank = c(2, 3, 5, 6, 11), seed = 123456)どのランクにするかについては複数の選び方が提唱されている。cophenetic が減少し始める時のランク、あるいは RRS 曲線の反曲点にあたるときのランクなどを選ぶ。
References
- Gene Expression Data Classification Based on Non-negative Matrix Factorization. IEEE. 2009. IEEE
- A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010, 11:367. PubMed Abstract CRAN