RNA-Seq のサンプルが複数あるとき、サンプル同士が似てるかどうかをあらかじめ調べておくことで、サンプル同士の関係性が明確になったり、外れ値的なサンプルが存在していることを確認できたりして、後々の解析の見通しがよくなる。サンプル同士の関係性をみるために、主成分分析や階層クラスタリングなどがある。これ以外に、多次元尺度構成法(MDS)も使われたりする。多次元尺度構成法は、複数のサンプルで計算された距離行列を、決められた手順にしたがって、低次元空間(例えば 2 次元空間)に変換する方法である。多次元尺度構成法で得られる結果は、クラスタリングの結果と似ているが、多次元尺度構成法はクラスタリングの方法ではない。
サンプルデータ
MDS で解析するサンプルデータを以下のようなものを用いる。A 群と B 群の 2 つの実験群からなる。A 群には薬剤 A を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。B 群には薬剤 B を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。各実験の各時刻において 2 つの biological replicate があり、実験全体で計 12 biological replicate を使用したとする。また、全遺伝子数を 1,000 とした。サンプルデータを読み込んでから行列型に変換し、count 変数に代入する。
count <- read.table("https://bi.biopapyrus.jp/data/counts_6_6_timecourse.txt", sep = "\t", header = T, row.names = 1)
count <- as.matrix(count)
dim(count)
## [1] 1000 12
head(count)
## A1_0h A2_0h A3_3h A4_3h A5_6h A6_6h B1_0h B2_0h B3_3h B4_3h B5_6h B6_6h
## gene_1 1 7 11 0 22 24 0 0 9 3 22 16
## gene_2 170 182 744 683 918 637 128 112 645 517 1248 1234
## gene_3 8 12 37 46 100 94 9 8 30 46 47 63
## gene_4 44 38 109 187 354 225 40 97 103 146 505 343
## gene_5 0 0 5 0 4 10 0 0 1 0 7 4
## gene_6 88 112 372 459 1010 611 129 80 486 360 678 767多次元尺度構成法 MDS
MDS を行うにあたって、ライブラリー間の距離行列を作成する必要がある。この距離行列は、スピアマンの順位相関係数を利用したり、線形回帰の決定係数を利用したりすることができる。スピアマンの順位相関係数を利用する場合は、以下のようにする。
rho <- cor(count, method = "spearman")スピアマンの順位相関係数は 0 ≤ ρ ≤ 1 の範囲内の値をとる。2 つのライブラリー同士が似ていれば相関係数は 1 に近い値をとり、似ていなければ相関係数は 0 に近い値をとる。この相関係数を距離として利用したいので、次のように、1 で相関係数を引く。この操作により、2 つのライブラリー同士が似ていれば距離は 0 に近い値をとり、似ていなければ距離は 1 に近い値をとるようになる。
d <- dist(1 - rho)
d
## A1_0h A2_0h A3_3h A4_3h A5_6h A6_6h B1_0h B2_0h B3_3h B4_3h B5_6h
## A2_0h 0.1460712
## A3_3h 0.1926113 0.1875459
## A4_3h 0.1879476 0.1939270 0.1469633
## A5_6h 0.1976550 0.1971289 0.1799684 0.1662711
## A6_6h 0.2105574 0.2106745 0.1771021 0.1782299 0.1519325
## ...距離行列 d に対して、MDS を行い、その結果をプロットする(以下の図は ggplot2 によるもの)。
mds <- cmdscale(d)
plot(mds, type = "n")
text(mds, labels = colnames(count))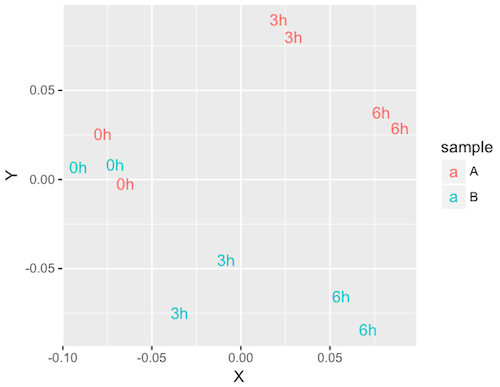
図から、A 群 0 hr と B 群 0 hr が、乱数生成条件と同じく、互いに近くなっていることが確認できる。また、3 hr、6 hr と時間がたつと、A 群と B 群が異なる方向へ離れていくのも確認できる。このように MDS で描いた図は、2 つのライブラリーが似ていれば、近くにプロットされる。また、MDS は距離行列を低次元空間に変換しているだけなので、図で表示されている横軸と縦軸は、特別な意味を持っていない。