サンプル同士の類似度を調べる際に、方法が簡単で、結果が解釈しやすい方法として階層クラスタリングがあげられる。ここで乱数の生成により作成したデータを利用してクラスタリングを行ってみる。
サンプルデータ
サンプルデータは条件を設定して負の二項分布に従うように乱数生成により作成した。A 群と B 群の 2 つの実験群からなる。A 群には薬剤 A を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。B 群には薬剤 B を投与し、0 時間、3 時間、6 時間の 3 つの時刻においてサンプルを採取した。各実験の各時刻において 2 つの biological replicate があり、実験全体で計 12 biological replicate を使用したとする。また、全遺伝子数を 1,000 とした。
サンプルデータを読み込んでから行列型に変換し、count 変数に代入する。
count <- read.table("https://bi.biopapyrus.jp/data/counts_6_6_timecourse.txt", sep = "\t", header = T, row.names = 1)
count <- as.matrix(count)
dim(count)
## [1] 1000 12
head(count)
## A1_0h A2_0h A3_3h A4_3h A5_6h A6_6h B1_0h B2_0h B3_3h B4_3h B5_6h B6_6h
## gene_1 1 7 11 0 22 24 0 0 9 3 22 16
## gene_2 170 182 744 683 918 637 128 112 645 517 1248 1234
## gene_3 8 12 37 46 100 94 9 8 30 46 47 63
## gene_4 44 38 109 187 354 225 40 97 103 146 505 343
## gene_5 0 0 5 0 4 10 0 0 1 0 7 4
## gene_6 88 112 372 459 1010 611 129 80 486 360 678 767一般的に、これらのデータを正規化した上で、分散の大きい遺伝子のみに対して、階層クラスタリングを行う。ここで、簡略化のため、正規化しないですべての遺伝子に対して階層クラスタリングを行う。
サンプル間の階層クラスタリング
まず、サンプル同士でどれぐらい似ているかを調べる。これにはスピアマンの順位相関係数を利用する。
rho <- cor(count, method = "spearman")相関係数が 1 に近ければ、サンプル同士が似ているということを示す。そこで、as.dist 関数で「1 - 相関係数」を距離として定義してクラスタリングを行う。クラスタリングアルゴリズムは、最大距離法や群平均法など様々あるが、比較的にウォード法が安定していて、よく利用される。
d <- as.dist(1 - rho)
h <- hclust(d, method = "ward")クラスタリングの結果を図示すると次のようになる。薬剤投与前(0 時間)の A 群と B 群は同じクラスターに入っている。薬剤投与後は、 A 群と B 群は、それぞれ異なるクラスターを形成した。また、薬剤投与後の A 群と B 群のクラスターの中には、時間に応じてさらにサブクラスターが形成された。
plot(h)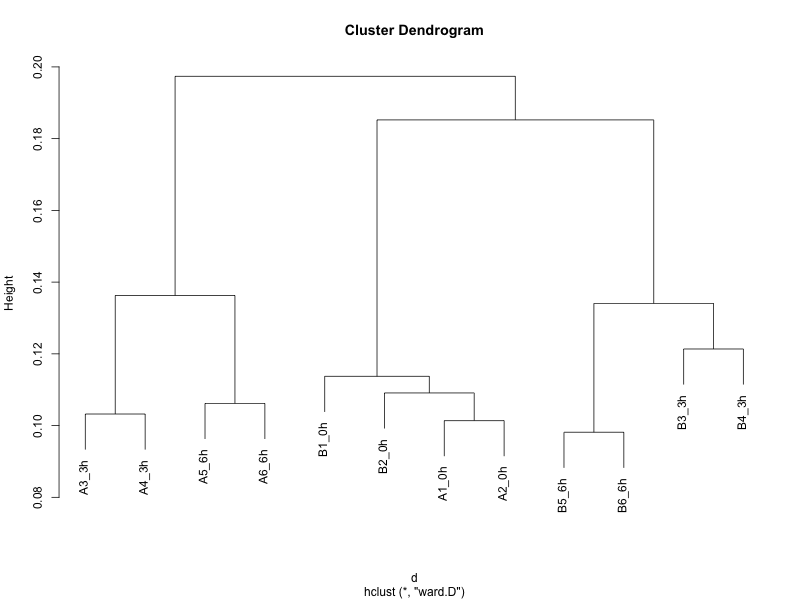
遺伝子間の階層クラスタリング
同じ発現パターンの遺伝子を見つけることが目的であれば、遺伝子間のクラスタリングを行うこともできる。クラスタリング手順は上述と同じだが、入力する遺伝子の発現量行列の行と列を入れ替える必要がある。
rho <- cor(t(count), method = "spearman")
d <- as.dist(1 - rho)
h <- hclust(d, method = "ward")
plot(h, labels = F, h = -1)