MODELLER の secondary_structure.alpha メソッドを利用することで、アミノ酸の一次配列から、α-ヘリックスを作成することができる。作成手順としては、以下のようにステップ分けできる。例に用いる一次配列を、1ALK の一部「DKQDHAANPCGQIGETVDLDEAVQRALE」とする。この配列は 1ALK の中で実際に α-ヘリックスである。
- 一次配列のモデルを作成する(build_sequence)
- タンパク質が取りうる構造を計算する(restraints.make)
- α-ヘリックスを作成する(secondary_structure.alpha)
- 構造を最適化する(optimize)
Python スクリプトは以下のようになる。
from modeller import *
from modeller.optimizers import conjugate_gradients
env = environ()
env.libs.topology.read('${LIB}/top_heav.lib')
env.libs.parameters.read('${LIB}/par.lib')
# 1.
m = model(env)
m.build_sequence('DKQDHAANPCGQIGETVDLDEAVQRALE')
# 2.
allatoms = selection(m)
m.restraints.make(allatoms, restraint_type='STEREO', spline_on_site=False)
# 3.
m.restraints.add(secondary_structure.alpha(m.residues))
# 4.
cg = conjugate_gradients()
cg.optimize(allatoms, max_iterations=100)
# save to PDB format
m.write(file='alpha-helix.pdb')Python スクリプトを make_alphahelix.py として保存し、MODELLER で実行すれば、結果として alpha-helix.pdb ファイルが生成される。
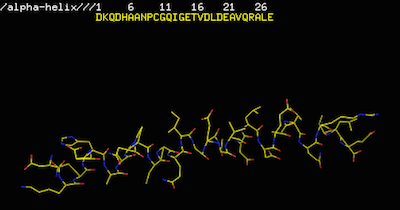
mod9.13 make_alphahelix.py作成された α-ヘリックスを PyMOL で見ると、以下のようになる。
メソッドの説明
スクリプト中で利用したいくつかのメソッドは、以下のような機能を持つ。
restraints.make |
配列上の各アミノ酸が取りうる構造を計算するメソッドである(と思う・・・)。計算アルゴリズムは restraint_type で指定できる。上の例では STEREO と指定したが、BOND, ANGLE, DIHEDRAL, IMPROPER, LJ14 など様々なアルゴリズムを指定することができる(詳細)。 |
secondary_structure.alpha |
二次構造を強制的に α-ヘリックスに構築させる(詳細)。 |
conjugate_gradients |
構造を最適化するための最適化オブジェクトを作成する(詳細)。 |
optimize |
MD により構造を最適化する。 |