pathview は R / Bioconductor パッケージで、KEGG パスウェイを視覚化できる。pathview を実行すると、必要なパスウェイの情報を HTTP あるいは API を通して KEGG からダウンロードする。pathview を利用するには、遺伝子 ID と KEGG パスウェイ ID が必要。
library(pathview)利用できる生物種
KEGG では 3,000 種以上の生物種のデータが保存されている。pathview パッケージを通して、利用できる生物種を確認するには以下のようにする。1 列目に表示される kegg.code が、生物種の指定に利用する。例えば、ヒトのデータを利用したい場合は、pathview(species = "hsa", ...) と指定すれば良い。
data(korg)
head(korg)
## kegg.code scientific.name common.name entrez.gnodes kegg.geneid ncbi.geneid
## [1,] "hsa" "Homo sapiens" "human" "1" "100" "100"
## [2,] "ptr" "Pan troglodytes" "chimpanzee" "1" "100533953" "100533953"
## [3,] "pps" "Pan paniscus" "bonobo" "1" "100967419" "100967419"
## [4,] "ggo" "Gorilla gorilla gorilla" "western lowland gorilla" "1" "101123859" "101123859"
## [5,] "pon" "Pongo abelii" "Sumatran orangutan" "1" "100169736" "100169736"
## [6,] "mcc" "Macaca mulatta" "rhesus monkey" "1" "100301991" "100301991"代謝パスウェイの描き方(A. Thaliana)
pathview 関数に遺伝子 ID とパスウェイの ID を指定すると、対応するパスウェイの図と KEGG XML データが自動的に KEGG ウェブサイトからダウンロードされる。ここで、シロイヌナズナの ath00460 パスウェイ(KEGG PATHWAY)を描く。このパスウェイには複数の遺伝子が登録されているが、ここでは 4 つの遺伝子(AT5G42260, AT5G54570, AT3G62750, AT5G05260)だけをマークアップする。ここで、とくに注意したいのは pathview を実行するときに、gene.idtype = "KEGG" を指定している。これを指定しないと、TAIR ID を識別してくれない。
gene.id <- c("AT5G42260", "AT5G54570", "AT3G62750", "AT5G05260")
path.id <- "00460"
pv <- pathview(gene.data = gene.id, pathway.id = path.id, species = "ath", gene.idtype = "KEGG")R を実行しているディレクトリに ath00460.png と ath00460.xml がダウンロードされ、また ath04600.pathview.png が作成される。
ath00460.png は KEGG ウェブサイトで公開されている画像と同じものになっている。
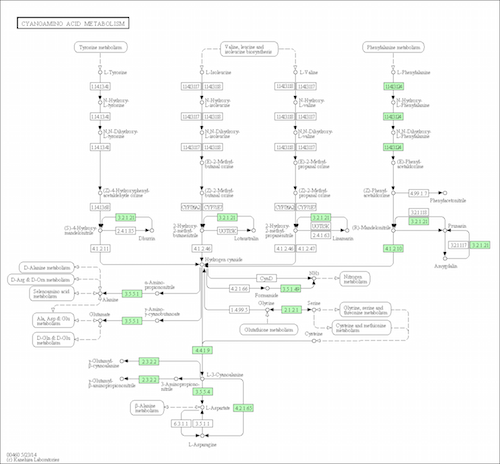
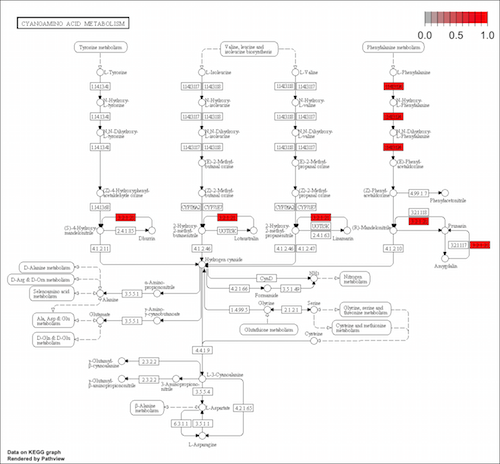
ath00460.pathview.png は KEGG ウェブサイトで公開されている画像に、gene.data に与えた遺伝子をマークアップした図になる。この例では赤く塗られている。
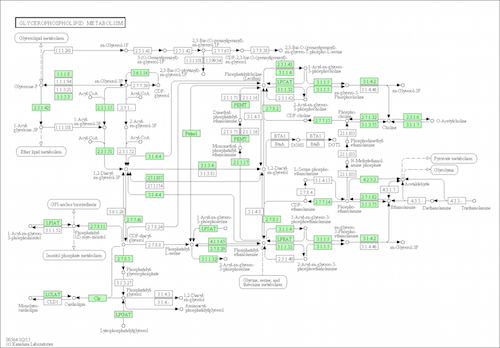
代謝パスウェイと化合物(H. sapiens)
次にヒトの例を取り上げる。人の場合は Entrez Gene ID を利用する(KEGG PATHWAY)。
gene.id <- c("2819", "2820", "8611", "100137049")
path.id <- "00564"
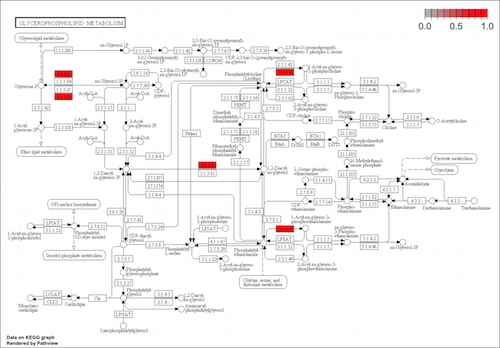
pv <- pathview(gene.data = gene.id, pathway.id = path.id, species = "hsa")この例では hsa000564.xml, hsa000564.png に加え hsa000564.patview.png も出力された。hsa000564.pathview.png の赤色に塗られている部分が入力遺伝子(genen.id)が関与している部分である。
パスウェイを描くためのデータは pv に保存されている。
head(pv$plot.data.gene)
## kegg.names labels type x y width height mol.data mol.col
## 70 81490 PTDSS2 gene 562 643 46 17 NA #FFFFFF
## 71 9791 PTDSS1 gene 495 345 46 17 NA #FFFFFF
## 81 8443 GNPAT gene 106 278 46 17 NA #FFFFFF
## 85 10162 LPCAT3 gene 737 211 46 17 NA #FFFFFF
## 86 10390 CEPT1 gene 702 280 46 17 NA #FFFFFF
## 87 5130 PCYT1A gene 835 311 46 17 NA #FFFFFF
pv$plot.data.gene[pv$plot.data.gene[, 9] == "#FF0000", ]
## kegg.names labels type x y width height mol.data mol.col
## 95 100137049 PLA2G4B gene 737 187 46 17 1 #FF0000
## 98 8611 PPAP2A gene 536 427 46 17 1 #FF0000
## 104 100137049 PLA2G4B gene 737 592 46 17 1 #FF0000
## 114 2819 GPD1 gene 164 191 46 17 1 #FF0000
## 116 2820 GPD2 gene 164 250 46 17 1 #FF0000
head(pv$plot.data.cpd)
## kegg.names labels type x y width height kegg.names mol.col
## 75 C06771 C06771 compound 1227 519 8 8 NA #FFFFFF
## 76 C06772 C06772 compound 1118 519 8 8 NA #FFFFFF
## 77 C00084 C00084 compound 1042 464 8 8 NA #FFFFFF
## 149 C00641 C00641 compound 682 437 8 8 NA #FFFFFF
## 150 C01996 C01996 compound 1118 312 8 8 NA #FFFFFF
## 151 C04230 C04230 compound 790 176 8 8 NA #FFFFFF次に、同じくヒトを例にして、遺伝子とともに化合物を与えてパスウェイを描く。
gene.id <- c("2819", "2820", "8611", "100137049")
path.id <- "00564"
cpd.id <- c("C00040", "C00084", "C00093")
pv <- pathview(gene.data = gene.id, cpd.data = cpd.id, pathway.id = path.id, species = "hsa")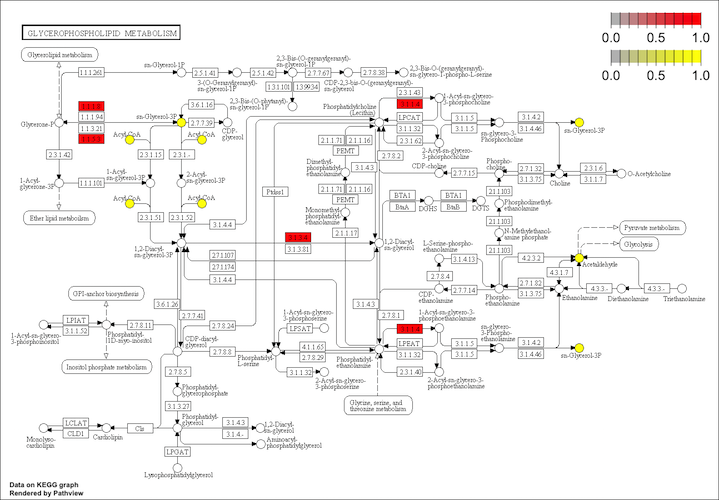
fold-change の表示
gene.data に遺伝子の名前を与える代わりに、名前付きの実数ベクトルを与えると、pathview はその実数値(fold-change)に基づいて、アノテーション遺伝子を色付けする。
gene.id <- c(2, 1.4, 0.3, -2) # log2 fold-change
names(gene.id) <- c("2819", "2820", "8611", "100137049")
path.id <- "00564"
pv <- pathview(gene.data = gene.id, pathway.id = path.id, species = "hsa")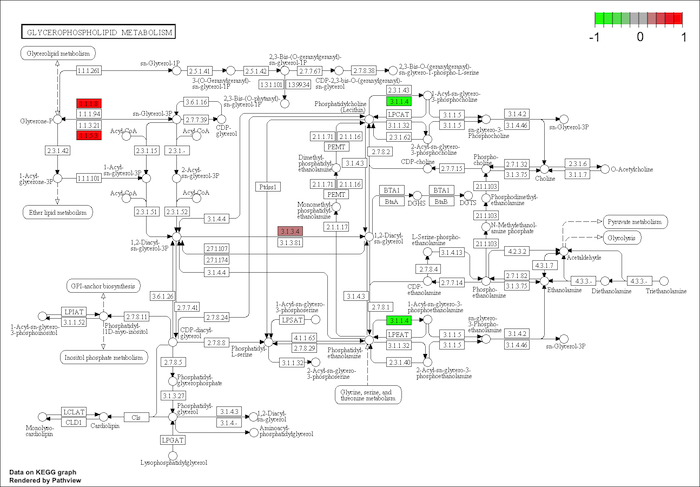
gene.id を行列型にし、行名を遺伝子名にすると、複数の fold-change を同時に色づけることができる。例えば、3 列の行列を利用した時は以下のようになる。時系列データの視覚化に便利。
gene.id <- matrix(c(1, 0.1, 0.3, -2, 0.5, 0.7, 1, 1, -0.5, 1, -0.7, -1), ncol = 3)
gene.id
## [,1] [,2] [,3]
## 2819 1.0 0.5 -0.5
## 2820 0.1 0.7 1.0
## 8611 0.3 1.0 -0.7
## 100137049 -2.0 1.0 -1.0
rownames(gene.id) <- c("2819", "2820", "8611", "100137049")
path.id <- "00564"
pv <- pathview(gene.data = gene.id, pathway.id = path.id, species = "hsa")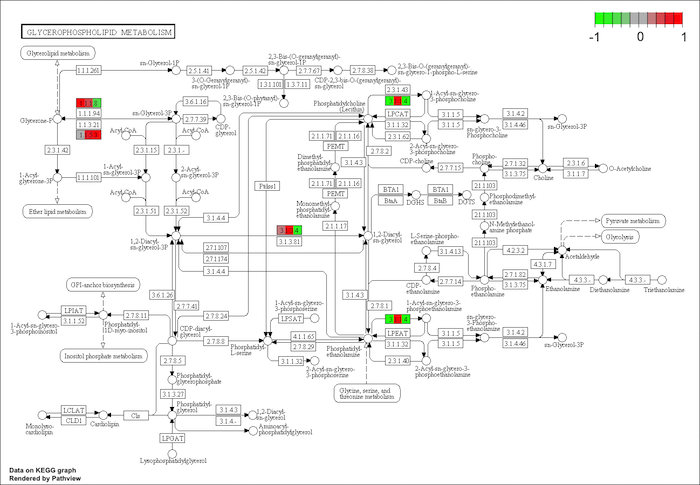
for 文を利用して複数のパスウェイマップの描き方
gene.data あるいは cpd.data には複数の遺伝子 ID と化合物 ID を代入できるが、pathway.id には複数の KEGG ID を代入すると、処理できないようである。そのため、複数のパスウェイのマップを作成したいときは、for で回すといい。
gene.id <- c("AT5G66120", "AT5G65770", "AT5G65780")
path.id <- c("00400", "00270", "00280")
pv <- vector("list", length(path.id))
for (i in 1:length(path.id)) {
pv[[i]] <- pathview(gene.data = gene.id, pathway.id = path.id[i], species = "ath", gene.idtype = "KEGG")
}