トランスクリプトーム解析による発現変動遺伝子を同定した後に、続けて GO 解析を行う場合がある。これは発現変動遺伝子だけが持つ GO terms が、全遺伝子の持つ GO terms に比べ、どれぐらい異なっているのかを調べるというものである。
topGO パッケージ(Bioconductor)ではこのような GO 解析の機能を提供している。このパッケージを利用して GO 解析するには以下のように行う。
- GO 解析用のデータを準備
発現変動遺伝子に対して GO 解析を行うのであれば、その遺伝子名(Ensembl ID など)、p-value あるいは FDR などを準備する。また、解析する生物種に対応したアノテーションデータベースもロードする。 - GO 解析
検定方法を指定して、GO term の検定を行う。 - GO 解析結果の解釈
検定結果を表に出力し、あるいはグラフとして出力し、有意差を持つ GO term について解釈を行う。
以下は topGO パッケージの利用例である。使用するサンプルデータは 2 列からなり、1 列目は遺伝子の Ensembl ID であり、2 列目は edgeR などのパッケージにより発現変動遺伝子を同定した結果として得られる p-value である。
# データを読み込む(1 列目は Ensembl ID、2 列目は p-value)
x <- read.table("https://bi.biopapyrus.jp/https://stats.biopapyrus.jp/data/counts_3_3.txt")
# p-value から FDR を計算する
fdr <- p.adjust(x[, 2], method = "BH")
names(fdr) <- x[,1]
# DEG 判定関数(FDR < 0.01 ならば DEG とする)
f <- function(q) {
return (q < 0.01)
}
# 解析準備
topgo <- new("topGOdata",
ontology = "CC", # オントロジー
allGenes = fdr, # FDR 値
geneSel = f, # DEG 判定関数
annot = annFUN.org, # アノテーション
mapping = "org.Hs.eg.db", # アノテーションデータ
ID = "Ensembl") # Ensembl ID を利用しているのでこれを指定
# GO term 検定
result <- runTest(topgo, algorithm = "classic", statistic = "fisher")
# GO term 検定結果を表に出力する(検定結果のトップ 10)
result.table <- GenTable(topgo, FisherTest = result, topNodes = 10)
result.table
## GO.ID Term Annotated Significant Expected FisherTest
## 1 GO:0005576 extracellular region 1153 105 52.08 2.4e-13
## 2 GO:0031674 I band 56 12 2.53 5.6e-06
## 3 GO:0030017 sarcomere 88 15 3.98 7.9e-06
## 4 GO:0030016 myofibril 103 16 4.65 1.3e-05
## 5 GO:0044449 contractile fiber part 97 15 4.38 2.6e-05
## 6 GO:0043292 contractile fiber 110 16 4.97 3.1e-05
## 7 GO:0001520 outer dense fiber 6 4 0.27 5.7e-05
## 8 GO:0097223 sperm part 51 10 2.30 7.6e-05
## 9 GO:0030018 Z disc 45 9 2.03 0.00015
## 10 GO:0031983 vesicle lumen 39 8 1.76 0.00029
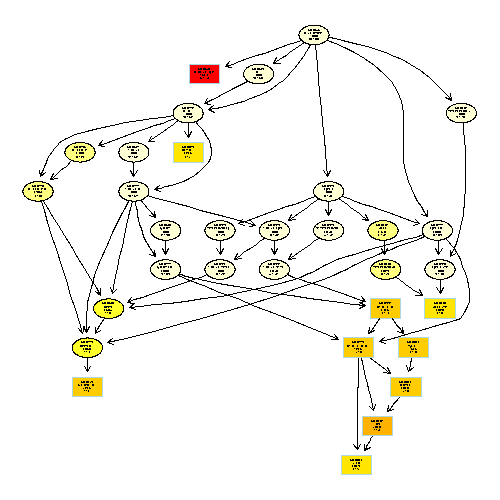
# GO term 検定結果のトップ 10 のネットワークを描く
showSigOfNodes(topgo,
score(result), # Fisher 検定の結果を利用する
firstSigNodes = 10, # Fisher 検定の結果のうち、有意差のもっとも大きい 10 ノードだけをプロットする
useInfo = "all")
実際に描かれるグラフは以下のようになる。firstSigNodes を 10 に指定しているため、実際に描かれるノード(四角)が 10 個となる。
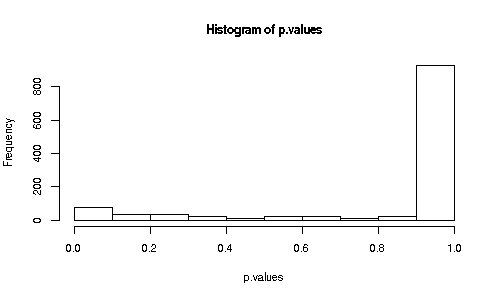
検定結果の分布を確認するにはヒストグラムを描く。検定結果は result に保存されているので、これを score 関数に与えて、統計量だけ取得し、ヒストグラムに描く。
p.values <- score(result) hist(p.values)
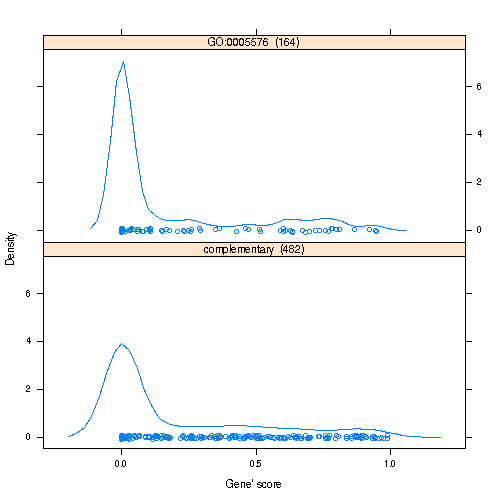
次に、個々の GO term がどのように分布しているのかを確認する。上例の続きとして、result.table 中の最初の GO term について、その分布を確認する。
goid <- result.table[1, "GO.ID"] # 1 番目の GO term (もっとも有意な GO term) print(showGroupDensity(topgo, goid)) goid ## [1] "GO:0005576"
グラフは以下のようになる。縦軸は推定密度を表し、出現頻度を割合で表したものである。横軸は Gene score であり、この場合 GO 解析に FDR を用いるので、Gene score は FDR となる。 2 つの密度曲線のうち、上側のグラフは GO:0005576 を持つ遺伝子を表している。164個の遺伝子がこの GO term を持ち、その多くが非常に小さい FDR を持つことがわかる。 一方、下側のグラフは全遺伝子のうち GO:0005576 を持たない遺伝子の分布を表している。上下のグラフを合わせて解釈すると、GO:0005576 は有意差を持つとは意でない。 すなわち Fisher 検定の結果でトップにランクされても、実際には有意でない可能性も含まれる。
他の検定法も合わせて検討する。
topgo <- new("topGOdata",
ontology = "CC", # オントロジー
allGenes = fdr, # FDR 値
geneSel = f, # DEG 判定関数
annot = annFUN.org, # アノテーション
mapping = "org.Hs.eg.db", # アノテーションデータ
ID = "Ensembl") # Ensembl ID を利用しているのでこれを指定
# GO term 検定
resultFisher <- runTest(topgo, algorithm = "classic", statistic = "fisher")
resultKS <- runTest(topgo, algorithm = "classic", statistic = "ks")
resultT <- runTest(topgo, algorithm = "classic", statistic = "t")
# GO term 検定結果を表に出力する(検定結果のトップ 10)
result.table <- GenTable(topgo,
Fisher = resultFisher,
KS = resultKS,
T = resultT,
orderBy = "KS", # 直前に定義した Fisehr, KS, T のいずれかを与える
topNodes = 10)
result.table
## GO.ID Term Annotated Significant Expected Rank in KS Fisher KS T
## 1 GO:0005737 cytoplasm 4866 172 219.81 1 1 7.4e-11 1
## 2 GO:0044422 organelle part 3328 107 150.33 2 1 3.5e-10 1
## 3 GO:0044446 intracellular organelle part 3274 98 147.89 3 1 4.6e-10 1
## 4 GO:0031974 membrane-enclosed lumen 1408 26 63.60 4 1 1.2e-08 1
## 5 GO:0070013 intracellular organelle lumen 1353 22 61.12 5 1 2.5e-08 1
## 6 GO:0043233 organelle lumen 1380 26 62.34 6 1 3.1e-08 1
## 7 GO:0044444 cytoplasmic part 3627 120 163.84 7 1 4.2e-08 1
## 8 GO:0005829 cytosol 1291 36 58.32 8 1 7.7e-07 1
## 9 GO:0005576 extracellular region 1153 105 52.08 9 2.4e-13 3.5e-06 5.1e-13
## 10 GO:0044428 nuclear part 1249 13 56.42 10 1 4.5e-06 1
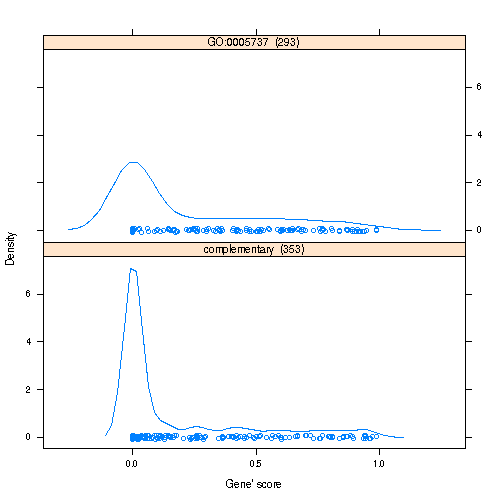
トップランクされた GO:0005737 の分布密度を描くと以下のようになる。(あまり変化してませんね。。。)
goid <- result.table[1, "GO.ID"] print(showGroupDensity(topgo, goid))