このページで ChIP-Seq データを解析する一連の流れを紹介する。Bowtie2、samtools、および MACS2 (Zhang et al.) を使用する。
データの準備
wget コマンドを使用して、ChIP-Seq の実験データ(FASTQ)を DDBJ からダウンロードする。ダウンロードした FASTQ ファイルは bzip2 形式で圧縮されているので、これらを展開する。ChIP-Seq データは Morey et al, 2013 のデータを利用する。
wget ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA061/SRA061964/SRX206418/SRR620204.fastq.bz2 #Ring1B_ChIPSeq
wget ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA061/SRA061964/SRX206419/SRR620205.fastq.bz2 #Cbx7_ChIPSeq
wget ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA061/SRA061964/SRX206420/SRR620206.fastq.bz2 #Suz12_ChIPSeq_2
wget ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA061/SRA061964/SRX206421/SRR620207.fastq.bz2 #RYBP_ChIPSeq
wget ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA061/SRA061964/SRX206422/SRR620208.fastq.bz2 #IgG_old_ChIPSeq
wget ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA061/SRA061964/SRX206423/SRR620209.fastq.bz2 #IgG_ChIPSeq_2
bunzip2 *.bz2本来は、ダウンロードしてきたデータに対してクオリティをチェックし、クオリティの低いリードを Trimmomatic などを使用して取り除いたりする。ここでは、この作業を省略する。
マッピング
マッピングの準備
FASTQ ファイル中のリードを、リファレンスゲノムにマッピングする。このデータはマウスのデータであるから、Ensembl からマウスのリファレンスゲノムをダウンロードしてくる。
wget ftp://ftp.ensembl.org/pub/release-91/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.toplevel.fa.gz
wget ftp://ftp.ensembl.org/pub/release-91/gtf/mus_musculus/Mus_musculus.GRCm38.91.gtf.gz
gunzip *.gzBowtie2 を使用して、リファレンスゲノムのインデックスを作成する。
bowtie2-build -f Mus_musculus.GRCm38.dna.toplevel.fa grcm38マッピング
Bowtie2 を使用してマッピングする。Bowtie2 のオプションは実験や生物に合わせて設定すればいい。ここでは、-3 5 --local とする。ユニークマッピング率は 40-62% であった。
bowtie2 -p 4 -3 5 --local -x grcm38 -U SRR620204.fastq -S ring1b.sam
bowtie2 -p 4 -3 5 --local -x grcm38 -U SRR620205.fastq -S cbx7.sam
bowtie2 -p 4 -3 5 --local -x grcm38 -U SRR620206.fastq -S suz12.sam
bowtie2 -p 4 -3 5 --local -x grcm38 -U SRR620207.fastq -S rybp.sam
bowtie2 -p 4 -3 5 --local -x grcm38 -U SRR620208.fastq -S igg_old.sam
bowtie2 -p 4 -3 5 --local -x grcm38 -U SRR620209.fastq -S igg.samマッピング結果は SAM ファイルに保存される。あとでピークコールするために、これらの SAM ファイルをソートして、BAM ファイルに変換する。
samtools sort -O bam -o ring1b.bam ring1b.sam
samtools sort -O bam -o cbx7.bam cbx7.sam
samtools sort -O bam -o suz12.bam suz12.sam
samtools sort -O bam -o rybp.bam rybp.sam
samtools sort -O bam -o igg_old.bam igg_old.sam
samtools sort -O bam -o igg.bam igg.samゲノムブラウザなどでマッピング結果をみるとき、ソートされた BAM ファイルのインデックスも必要である。ここで、すべての BAM ファイルについて、インデックスを作成する。
samtools index ring1b.bam
samtools index cbx7.bam
samtools index suz12.bam
samtools index rybp.bam
samtools index igg_old.bam
samtools index igg.bamピークコール
MACS2 を使用してピークコールを行う。コントロール群の BAM ファイルを -c 引数で指定し、処理群の BAM ファイルを -t 引数で指定する。また、ピークコールの際の閾値を -p または -q で指定する。ここでは、p-value < 10-5 と指定した。また、引数 -g にはゲノムサイズを指定するが、マウスの場合は単に -g mm とすればいい。-n は、任意の実験名を指定できる。
macs2 callpeak -c igg_old.bam -t suz12.bam -p 1e-5 -f BAM -g mm -n iggold_vs_suz12
macs2 callpeak -c igg_old.bam -t ring1b.bam -p 1e-5 -f BAM -g mm -n iggold_vs_ring1b
macs2 callpeak -c igg_old.bam -t cbx7.bam -p 1e-5 -f BAM -g mm -n iggold_vs_cbx7
macs2 callpeak -c igg_old.bam -t rybp.bam -p 1e-5 -f BAM -g mm -n iggold_vs_rybpMCAS2 によるピークコール後に、検出されたピークなどは iggold_vs_suz12_model.r、iggold_vs_suz12_peaks.narrowPeak、iggold_vs_suz12_peaks.xls、iggold_vs_suz12_summits.bed などのファイルに保存されている。出力ファイルに関して、詳細の説明は MACS2 の配布レポジトリに書かれている。
| _peaks.xls | タブ区切りのテキストファイル。染色体番号、ピーク開始位置、終了位置、ピーク頂点の位置、p-value などのデータが記載されている。Exxel などのアプリケーションで開くことができる。位置番号は 1 から数える。 |
| _peaks.narrowPeak | _peaks.xls とほぼ同一内容で、BED フォーマットのファイル。染色体番号、ピーク開始位置、終了位置などが記載されている。UCSC ゲノムブラウザで読み取ることができる。 |
| _summits.bed | BED フォーマット。各ピークの開始位置と終了位置は記載されていない。各ピークの頂点位置だけが記載されている。 |
| _model.r | ピークコールに用いたモデルを描く R コード。R でコードを実行すると、グラフが描かれる。 |
head iggold_vs_suz12_peaks.narrowPeak
## 1 3671223 3671442 iggold_vs_suz12_peak_1 123 . 6.26360 12.36445 8.95524 73
## 1 3672387 3672638 iggold_vs_suz12_peak_2 75 . 5.14896 7.59354 4.38918 25
## 1 4491585 4493792 iggold_vs_suz12_peak_3 659 . 24.70606 65.99286 59.97961 1029
## 1 4496123 4497665 iggold_vs_suz12_peak_4 266 . 10.81543 26.62870 22.65303 1025
## 1 5018493 5020933 iggold_vs_suz12_peak_5 69 . 3.01293 6.98403 3.80816 230
## 1 5916298 5917508 iggold_vs_suz12_peak_6 328 . 9.47399 32.81531 28.58707 527
## 1 6359140 6359451 iggold_vs_suz12_peak_7 126 . 6.82214 12.64594 9.22468 201
## 1 6382845 6383150 iggold_vs_suz12_peak_8 121 . 6.55153 12.18861 8.78552 220
## 1 9299647 9300190 iggold_vs_suz12_peak_9 185 . 9.67389 18.56398 14.91139 384
## 1 9545526 9546245 iggold_vs_suz12_peak_10 189 . 5.85678 18.95214 15.28237 209
head iggold_vs_suz12_summits.bed
## 1 3671296 3671297 iggold_vs_suz12_peak_1 12.36445
## 1 3672412 3672413 iggold_vs_suz12_peak_2 7.59354
## 1 4492614 4492615 iggold_vs_suz12_peak_3 65.99286
## 1 4497148 4497149 iggold_vs_suz12_peak_4 26.62870
## 1 5018723 5018724 iggold_vs_suz12_peak_5 6.98403
## 1 5916825 5916826 iggold_vs_suz12_peak_6 32.81531
## 1 6359341 6359342 iggold_vs_suz12_peak_7 12.64594
## 1 6383065 6383066 iggold_vs_suz12_peak_8 12.18861
## 1 9300031 9300032 iggold_vs_suz12_peak_9 18.56398
## 1 9545735 9545736 iggold_vs_suz12_peak_10 18.95214結果表示
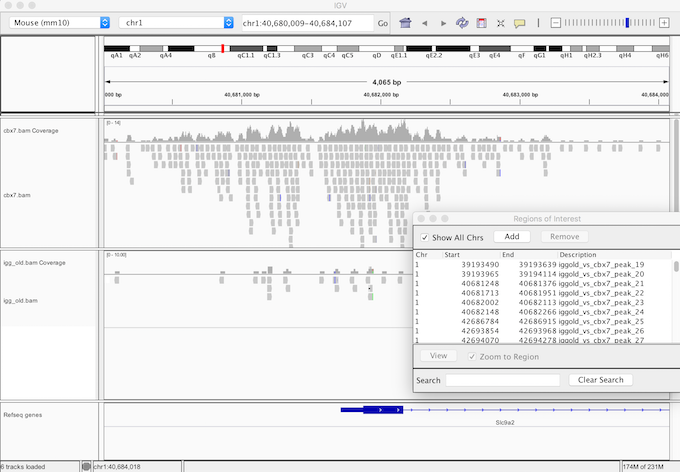
ピークコールの結果は IGV などのゲノムブラウザで視覚的に確認できる。IGV ゲノムブラウザでピークコールの結果を見る場合は、次のようにする。
- マウスゲノムを IGV に読み込む。
- IGV メニューバーから Genome > Create .genome file を選ぶ。
- Unique identifier と Descriptive name にはゲノムの名前などを記入する。
- FASTA の欄にはダウンロードした toplevel の FASTA ファイルを指定する。
- Optional の Gene file にはダウンロードした GFF ファイルを指定する。
- IGV で作成したゲノムを選択する。
- BAM ファイルを読み込む。
- IGV メニューバーから File > Load File を選ぶ。
- igg_old.bam を選ぶ。
- 同様にして、cbx7.bam を選ぶ。
- ピーク位置のデータを読み込む。
- IGV メニューバーから Regions > Import Regions を選ぶ。
- 次に、同じく Regions > Region Navigator を選ぶ。
- ナビゲーターウィンドウが立ち上がるので、ウィンドウ内に表示された各ピークの名前をクリックすると、IGV はそのピークの座標まで移動する。
References
- RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells. Cell Rep. 2013, 3(1):60-9. DOI: 10.1016/j.celrep.2012.11.026 PMID: 23273917
- Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9(9):R137. DOI: 10.1186/gb-2008-9-9-r137 PMID: 18798982