ChIP-Seq データのバイオインフォマティクス解析
ChIP-Seq
ChIP-Seq は、高速シーケンサーを利用して、タンパク質-DNA 相互作用部分を検出する技術である。DNA メチル化やヒストン修飾などのエピジェネティックな修飾や転写調節因子の結合部位を、ゲノムワイドスケールで網羅的に解析することができる。
ChIP-Seq 原理
核内の DNA は、転写因子をはじめとする様々なタンパク質と結合している。ChIP-Seq では、それらのタンパク質が DNA 上のどの部位に結合しているかを解明できる。原理的には、DNA と結合しいるタンパク質を架橋結合により固定したのちに、DNA を断片化する。続いて、このサンプル溶液に、解明したいタンパク質に特異的に結合する抗体を用いて、ターゲットタンパク質だけを沈殿させる。最後に、その沈殿を取り出して、架橋結合を壊し、DNA を精製してシーケンサーで読み取る。シーケンサーから出力された DNA 断片(リード)を全ゲノムにマッピングすれば、ターゲットタンパク質がどの部位に結合していたのかを明らかにすることができる。
ChIP-Seq データ解析
シーケンサーから出力される ChIP-Seq のデータは短い DNA 断片である。リードの配列を、全ゲノムの配列と照合し、リードがゲノム上のどの部分にアラインメントされるのかを調べる。この作業をマッピングとよぶ。マッピング後、ゲノム配列上のどの部位にリードが多くマッピングできたのかを調べる。そのような部位は、ターゲットタンパク質と結合(相互作用)している可能性が高いと考えられる。マッピング結果から DNA 結合部位を統計的に調べる作業をピークコールとよぶ。
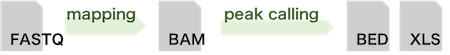
解析の流れ
ChIP-Seq データの解析は、マッピングまで RNA-Seq データの解析と同じ流れて行う。つまり、FASTQ ファイル中のリードに対して、クオリティチェックを行い、低品質のリードを除去する。続いて、リードをゲノム上マッピングする。ChIP-Seq のリードは、DNA の塩基配列をシーケンシングしたデータであるため、RNA-Seq のように、マッピング時にギャップを考慮する必要はない。マッピング後、ChIP-Seq 解析では、ピークコールを行う。続いて、ピークコールによって検出した部位がどの遺伝子に関係しているのかを調べていくアノテーション作業を行う。
ピークコール
タンパク質と結合している DNA 上の結合部位を統計的に調べる。ピークコールは、次のようなツールが用いられる。ツールごとに検出アルゴリズムが異なり、解析目的に応じて使い分ける必要がある。
| MACS | |
| FindPeak | |
| BroadPeak | |
| F-Seq | |
| SWA |
アノテーション
ピークコールによって得られた結合部位は、どの遺伝子の付近にあるのか、どのように遺伝子の発現を制御していくのかを調べる。
解析例
| Bowtie2 + MACS2 | マウス |