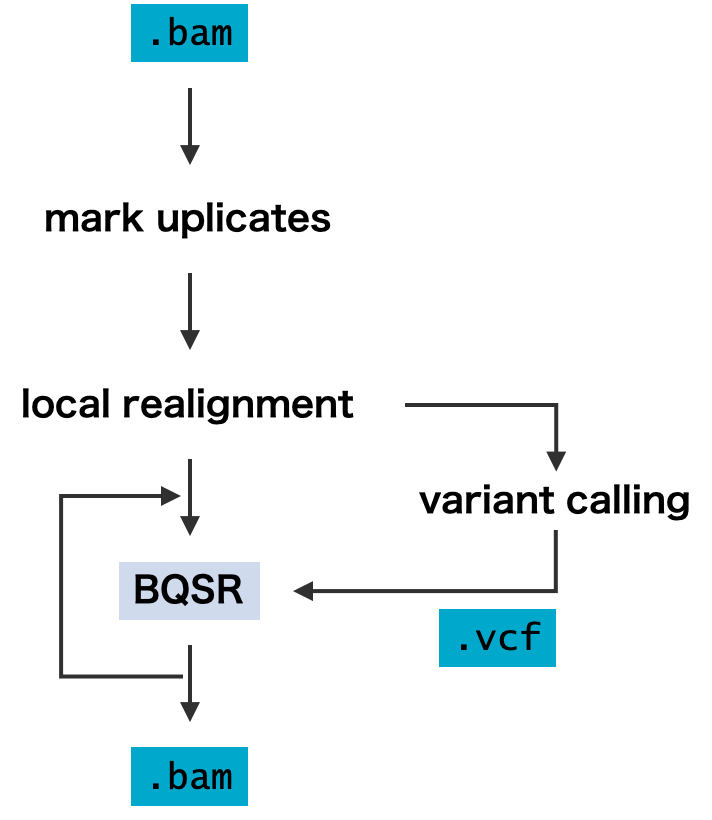
FASTQ ファイル中のクオリティスコアはシーケンサーの特有なエラーを含んだり、マッピング結果にはマッピングプログラムに特有なエラーを含んだりする。そのため、マッピングした直後の BAM ファイルを利用してそのまま variant calling を行うと、偽陽性を高める原因となる。そこで、variant calling を行う前に、マッピング結果の BAM ファイルに対して様々な補正を施す必要があり、これを前処理とよぶ。前処理は、重複リードの処理、再アラインメント、Base quality score recalibration (BQSR) とがある。
重複リードの処理
ライブラリーを準備する際に行われる PCR 増幅では、同一断片から複数のリードが生成される場合がある。また、シーケンシングにおけるコロニー形成のときに、あるコロニーがたまたま幅広く形成され、2 つのコロニーとして処理される場合がある。このように、ライブラリー中にある同一 DNA または RNA 断片から複数のリードが得られる場合がある。このような重複リードがマッピング結果に多く含まれると、重複リード上にたまたまシーケンサーエラーが存在した時に、その効果が拡張されて、それを間違って variant として同定してしまう危険性がある。これを防ぐために、重複リードを除去したり、あるいは目印をつけてあとで処理しないようにしておくことが大切である。
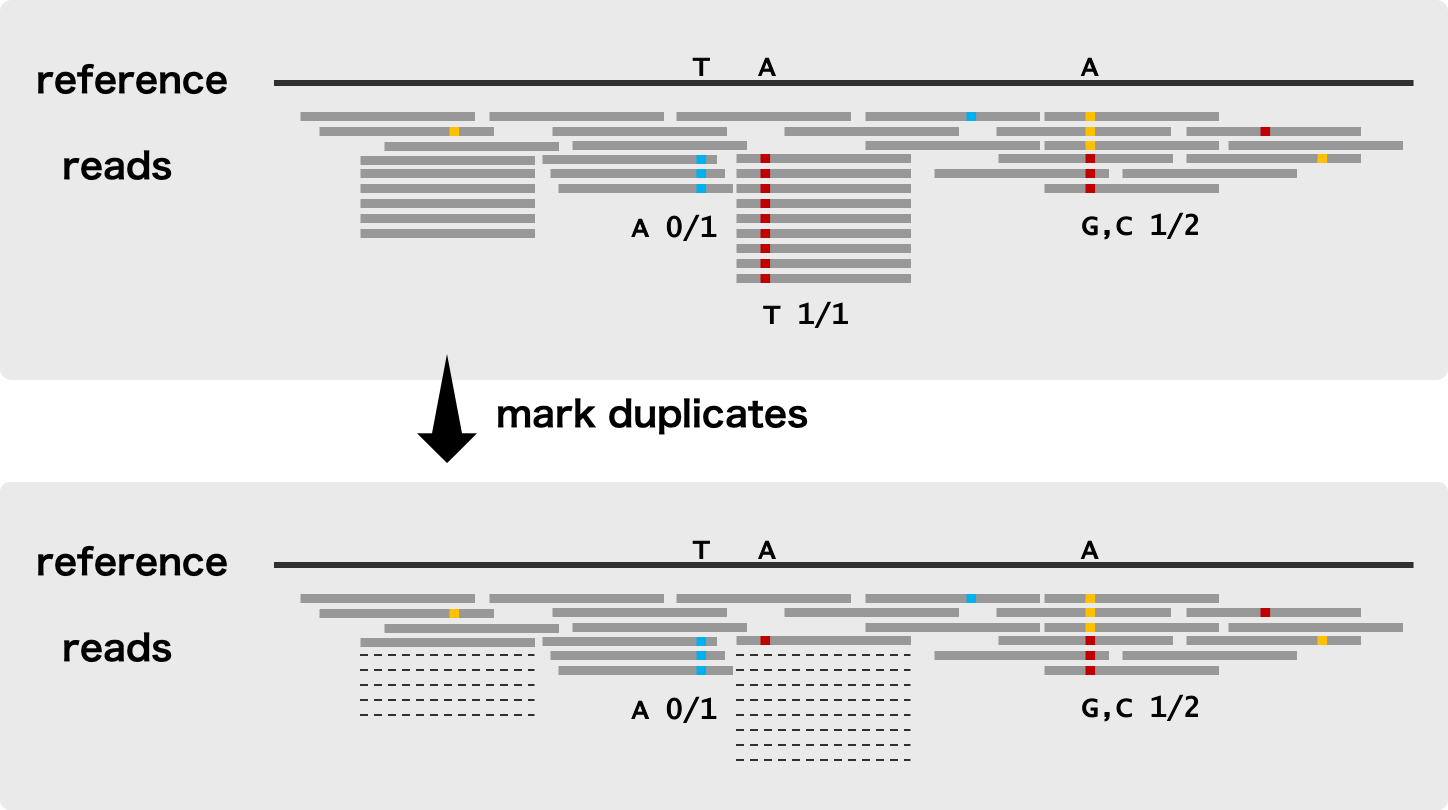
GATK では、MarkDuplicates コマンドを利用して重複リードに目印をつけることができる。このコマンドを使うことで、それぞれのリードが重複リードかどうかの情報が追加されて、新しい BAM ファイルとして保存される。このコマンドでは、重複リードが削除されることはない。
gatk MarkDuplicates -I input.bam -O output.bam -M markup_metrics.txtローカル再アラインメント
変異の入り方が複雑なとき、文字列検索アルゴリズムを使用したマッピングプログラムでは対応できない場合がある。この際に、変異の箇所を含めてその付近領域を Smith-Waterman アルゴリズムでアラインメントし直すことによって、正確なアラインメントが得られることが多い。
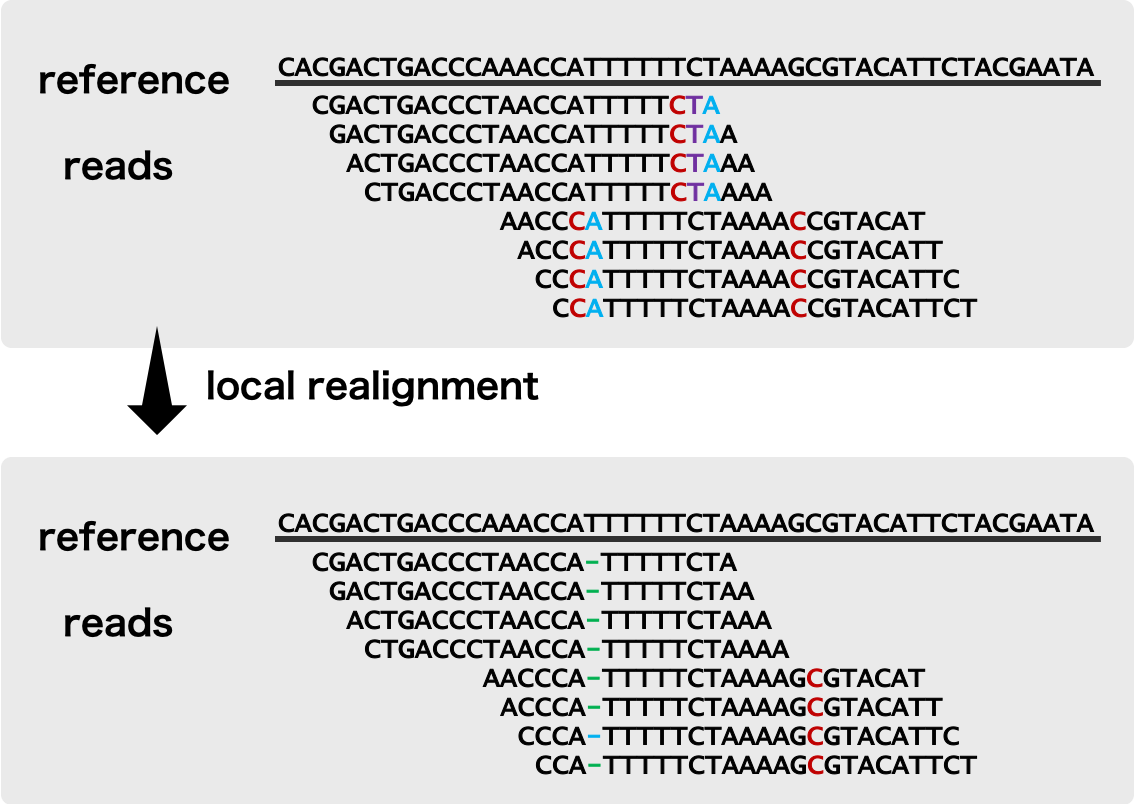
GATK3 では再アラインメントを行うには RealignerTargetCreator コマンドを利用する必要があったが、GATK4 からはこの機能が HaplotypeCaller コマンドに組み込まれるようになり、前処理として実行する必要がなくなった。
BQSR
FASTQ ファイル中に記載されたクオリティスコアは正確な値を反映していない場合がある。その理由として、一つにはこのスコアは PCR 時のエラーが含まれていない、もう一つはシーケンサーがこれらの値を出力するしくみ自体がブラックボックスとなっているためである。そこで、シーケンサーから得られるこれらのクオリティスコアが正確な値に近づくように補正する必要がある。この補正処理を Base Quality Score Recalibratio (BQSR) という。
BQSR は、既知の variants を除いて残った部分のアラインメント領域のデータを利用して、機械学習モデルを構築し、そのモデルを使って全体のクオリティスコアを補正する。そのため、BQSR を行うには、既知の variants 情報を必要とする。既知の variants 情報を入手できる場合はその情報を使用すればよいが、入手できない場合は自ら暫定な variants 情報を作成する必要がある。
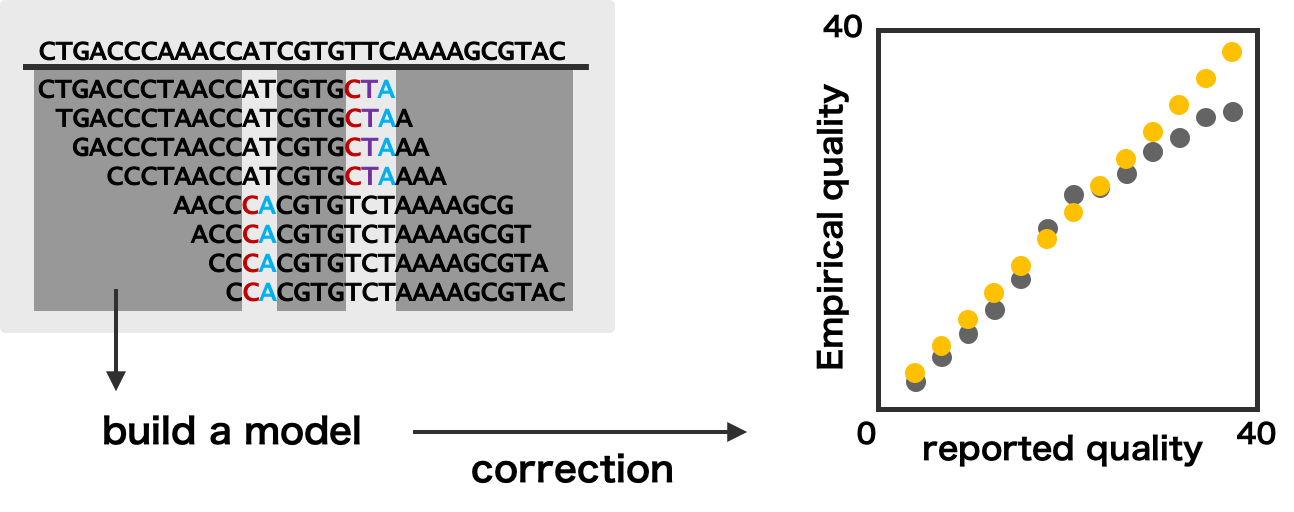
ヒトやマウスなどの生物では、既知の variants 情報がデータベースで保存されたり、あるいは先行研究としてまとめられている場合がある。それらの情報を vcf ファイルとして入手できれば、BQSR を次のように行うことができる。
gatk BaseRecalibrator -R genome.fa -I input.bam \
--known-sites dbsnp_146.b38.vcf.gz \
--known-sites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz \
--known-sites resources_broad_hg38_v0_1000G.phase3.integrated.sites_only.hg38.vcf \
-O output_recal_data.table
gatk ApplyBQSR -R genome.fa -I input.bam -bqsr output_recal_data.table -O output_bqsr.bam既知の variants 情報がない場合は、クオリティ補正していない BAM データから一度 variant calling を行い、ここで得られる信用性の低い variants (*.vcf)を既知情報として用いて BQSR を行う。
以上により、前処理は概ね次のような流れとなる。