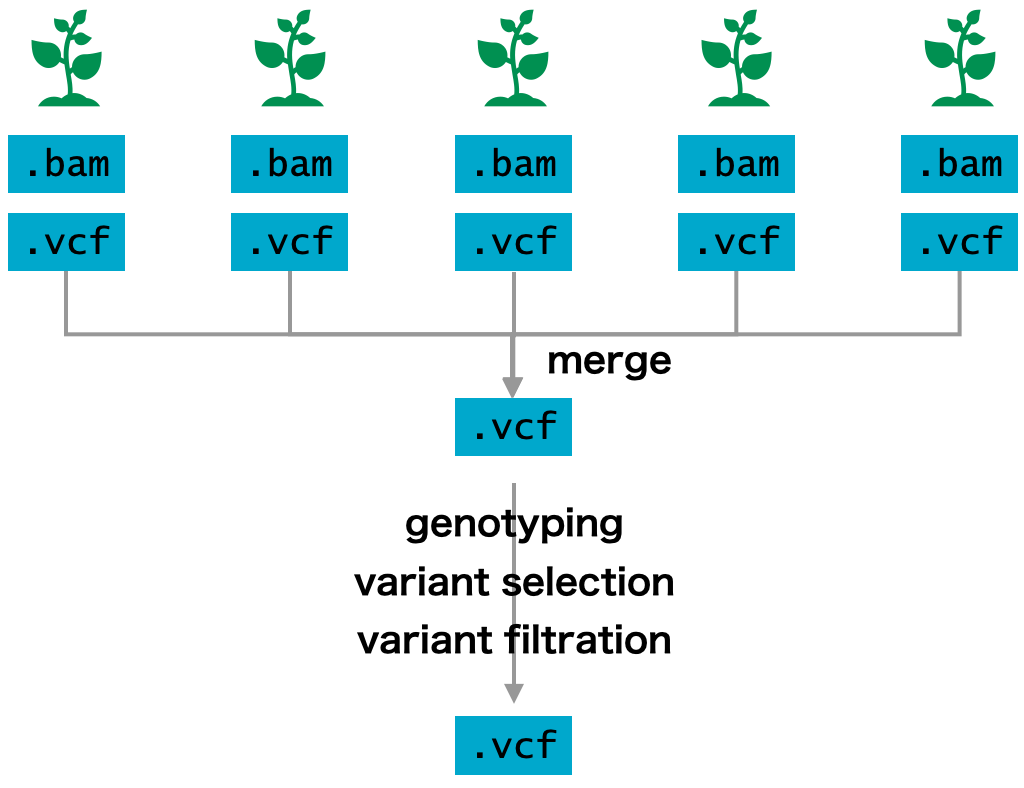
ジェノタイピングを行うとき、サンプルごとに行う single sample genotyping と、すべてのサンプルをまとめて行う joint genotyping とがある。single sample genotyping は 1 サンプルずつで処理を行うため、サンプルが取得次第解析できるため、結果を早く示すことができる。一方で、joint genotyping はすべてのサンプルが揃ってから一括に解析を行う必要があるため、新しいサンプルが追加されるたびに variant calling を行う必要がある。しかし、joint genotyping では、単一のサンプルではカバレッジ不足などが原因で variant として検出できなかった箇所も検出できるため、精度が高い。一般的に joint genotyping が推奨されている。
single sample genotyping
GATK では、single sample genotyping を行うのであれば、ハプロタイプの推定とジェノタイピングを同時に行うことができる。これらを行うコマンドは、HaplotypeCaller である。このコマンドにリファレンス配列(FASTA)、マッピング結果(BAM)を与えると、そのまま variant 情報の入った VCF を得ることができる。
gatk HaplotypeCaller -R genome.fa -I input.bam -O output.vcf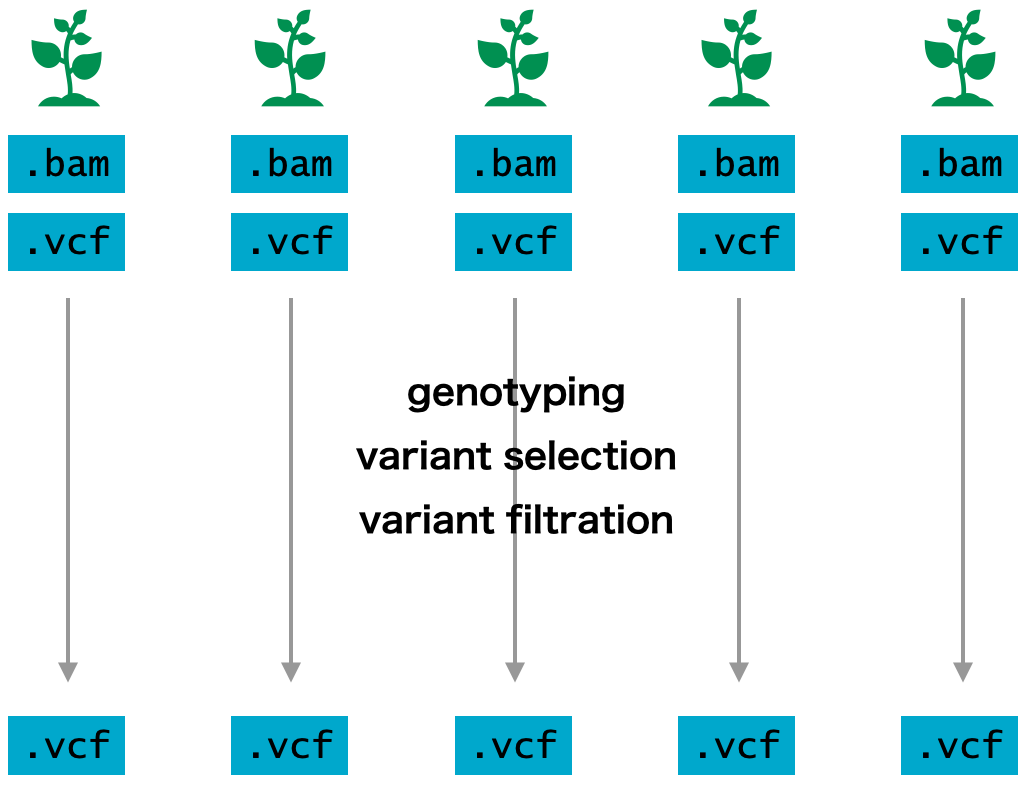
joint genotyping
joint genotyping では、複数のファイルをまとめてジェノタイピングを行う。joint genotyping を行うには、まず個々のサンプルに対してハプロタイプの推定を行い、その中間結果 gVCF を保存する。次に、それらの中間ファイルにある情報を 1 つのローカルデータベースとしてまとめる。最後に、そのローカルデータベースの情報を処理して、ジェノタイピングを行う。
ハプロタイプの推定は HaplotypeCaller コマンドを利用する。このとき、最後までジェノタイピングを行わずに、途中で止めて中間結果を保存するように --emit-ref-confidence GVCF オプションをつける。中間ファイルをローカルデータベースにまとめるときは GenomicsDBImport コマンドを利用する。この際に、対象染色体の番号あるいは特定の領域を指定する必要がある。1 つの領域だけを対象としたい場合は、-L Chr1 のように指定すればよい。複数の領域を対象としたい場合は、テキストファイルを用意し、1 行 1 領域のように情報を記入し、そのファイルパスを-L に与える。また、データベースからジェノタイピングを行うには GenotypeGVCFs コマンドを使用する。
gatk HaplotypeCaller -R genome.fa -I input_1.bam -O output_1.g.vcf --emit-ref-confidence GVCF
gatk HaplotypeCaller -R genome.fa -I input_2.bam -O output_2.g.vcf --emit-ref-confidence GVCF
echo -e "Chr1\nChr2\nChr3\nChr4\nChr5" > intervals.list
gatk GenomicsDBImport -R genome.fa -V output_1.g.vcf -V output_2.g.vcf -L intervals.list \
--genomicsdb-workspace-path gvcfs_db
gatk GenotypeGVCFs -R genome.fa -V gendb://gvcfs_db -O merged.vcf