CODEX(Jiang et al., 2015)は CNVs を検出する R/Bioconductor パッケージである。サンプルのデータは BAM フォーマットで与え、CNVs の検出範囲は BED フォーマットで与える。
以下に CODEX の使い方を示す。詳しい使い方は CODEX の vignette に書かれている。
まず、R を起動して、1000 ゲノムプロジェクトのゲノムデータをサンプルデータとして、R に取り込む。まず、BAM ファイルへのパスをベクトル形式で変数に保存する。各 BAM ファイルはあらかじめソートし、インデックス(.bai ファイル)を作成しておく必要がある。次に、サンプル名を 1 列の行列形式で変数に保存する。
library(CODEX)
library(WES.1KG.WUGSC)
# BAM files
dirPath <- system.file("extdata", package = "WES.1KG.WUGSC")
bamFile <- list.files(dirPath, pattern = '*.bam$')
bamdir <- file.path(dirPath, bamFile)
head(bamdir)
## [1] "/WES.1KG.WUGSC/extdata/NA06994.mapped.ILLUMINA.bwa.CEU.exome.20120522.bam.chr22.sorted.bam"
## [2] "/WES.1KG.WUGSC/extdata/NA10847.mapped.ILLUMINA.bwa.CEU.exome.20121211.bam.chr22.sorted.bam"
## [3] "/WES.1KG.WUGSC/extdata/NA11840.mapped.ILLUMINA.bwa.CEU.exome.20120522.bam.chr22.sorted.bam"
## [4] "/WES.1KG.WUGSC/extdata/NA12249.mapped.ILLUMINA.bwa.CEU.exome.20120522.bam.chr22.sorted.bam"
## [5] "/WES.1KG.WUGSC/extdata/NA12716.mapped.ILLUMINA.bwa.CEU.exome.20121211.bam.chr22.sorted.bam"
## [6] "/WES.1KG.WUGSC/extdata/NA12750.mapped.ILLUMINA.bwa.CEU.exome.20130415.bam.chr22.sorted.bam"
# sample names
sampname <- as.matrix(read.table(file.path(dirPath, "sampname")))
head(sampname)
## [1,] "NA06994"
## [2,] "NA10847"
## [3,] "NA11840"
## [4,] "NA12249"
## [5,] "NA12716"
## [6,] "NA12750"次に、CNVs の検出ターゲット領域を BED フォーマットで用意する。
bedFile <- file.path(dirPath, "chr22_400_to_500.bed")用意した BAM フォーマットのファイル、サンプルの名前、および BED フォーマットのファイルを getbambed 関数に与えて、解析用のオブジェクトを作成する。
chr <- 22
bambedObj <- getbambed(bamdir = bamdir, bedFile = bedFile,
sampname = sampname, projectname = "prjCNVs", chr = chr)BAM ファイルから sequence depth を計算する。Y には sequence depth が保存され、行は CNVs 検出のターゲット領域、列はサンプルを表す。
coverageObj <- getcoverage(bambedObj, mapqthres = 20) Y <- coverageObj$Y
GC 含量およびマッピング率を計算する。
gc <- getgc(chr, ref) mapp <- getmapp(chr, ref)
クオリティコントロールを行う。
qcObj <- qc(Y, sampname, chr, ref, mapp, gc, cov_thresh = c(20, 4000),
length_thresh = c(20, 2000), mapp_thresh = 0.9, gc_thresh = c(20, 80))
str(qcObj)
## List of 6
## $ Y_qc : int [1:77, 1:46] 52 25 57 101 149 95 50 79 181 100 ...
## $ sampname_qc: chr [1:46] "NA06994" "NA10847" "NA11840" "NA12249" ...
## $ gc_qc : num [1:77] 60.5 67.9 64.1 61.9 62.7 ...
## $ mapp_qc : num [1:77] 1 1 1 1 1 1 1 1 1 1 ...
## $ ref_qc :Formal class 'IRanges' [package "IRanges"] with 6 slots
## .. ..@ start : int [1:77] 21346453 21348163 21348797 21349109 21349985 21350935 21351471 21354119 21354886 21355495 ...
## .. ..@ width : int [1:77] 256 446 270 257 467 371 217 653 241 256 ...
## .. ..@ NAMES : NULL
## .. ..@ elementType : chr "integer"
## .. ..@ elementMetadata: NULL
## .. ..@ metadata : list()
## $ qcmat : num [1:100, 1:12] 22 22 22 22 22 22 22 22 22 22 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : NULL
## .. ..$ : chr [1:12] "chr" "start_bp" "end_bp" "pass" ...
ポアソンモデルにフィッティングさせ、sequencing depth の正規化を行う。パラメーター K を 1 から 9 まで試し、それぞれに対して AIC, BIC および RSS を計算し、最適な K を決定する。
normObj <- normalize(qcObj$Y_qc, qcObj$gc_qc, K = 1:9) Yhat <- normObj$Yhat AIC <- normObj$AIC BIC <- normObj$BIC RSS <- normObj$RSS K <- normObj$K par(mfrow = c(1, 3)) plot(K, RSS, type = "b", xlab = "Number of latent variables") plot(K, AIC, type = "b", xlab = "Number of latent variables") plot(K, BIC, type = "b", xlab = "Number of latent variables")
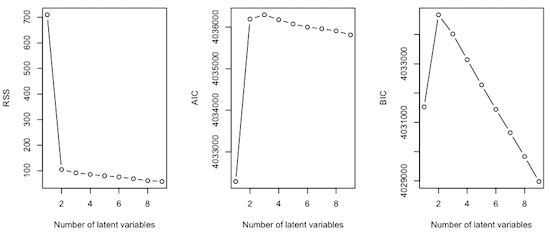
一般的に、AIC または BIC を指標として用いる場合は、AIC または BIC が最も大きくなる K を選ぶ。RSS を指標として用いる場合は、RSS が最も小さくなる K を選ぶ。ここでは、BIC が最も大きくなる K を用いて、CNVs 検出を行う。
optK < K[which.max(BIC)] # K = 2 in this case
finalcall <- segment(qcObj$Y_qc, Yhat, optK = optK, K = K, qcObj$sampname_qc,
qcObj$ref_qc, chr, lmax = 200, mode = "integer")
finalcall
## sample_name chr cnv st_bp ed_bp length_kb st_exon ed_exon raw_cov norm_cov copy_no lratio mBIC
## "NA18990" "22" "dup" "22312814" "22326373" "13.56" "60" "72" "1382" "1000" "3" "60.33" "49.728"
References
- CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 2015, 43(6):e39. DOI: 10.1093/nar/gku1363 PMID: 25618849